ChIP-exo Tutorial
Generating two basic sequence-specific ChIP-exo plots: composite and heatmap
Goal: This tutorial provides a guide to generating 2 basic plots (Composite plot and Heatmap) using the ScriptManager platform and data generated by the Yeast Epigenome project.
Download ScriptManager (v0.14):
The current version of ScriptManager is available for download here. Make sure you have Java installed.
The file ScriptManager-v0.14.jar should be placed someplace locally accessible. For example on Mac OS on the Desktop (Permissions will need to be accepted) or someplace in your home directory (Macintosh_HD:Users/userID/ScriptManager)
Download Data
You need one set of genomic coordinate regions to investigate (BED) and one file of sequencing data alignments (BAM) to complete this exercise. Read more about the BED/BAM file formats here.
BED File
This is the set of Reb1 binding sites from the Rossi et al (2018).
Download sample BED fileIf your BED file downloads with a .txt extension, make sure to change the filename to a .bed extension. For this tutorial, the BED file is named Reb1_Rhee_primary_sites_975.bed.
BAM File
This is the set of Reb1 read alignments from the Yeast Epigenome Project (YEP). See Rossi et al (2021) for more details.
Download sample BAM fileOR

- Navigate to www.yeastepigenome.org and search for Reb1
- Select "META DATA"
- Select "Direct Download"
- Unzip the resulting file ‘12141_YEP.zip’ and inspect the contents of the new
12141_YEPfolder. It should contain a file called12141_filtered.bam.
Generate the Plots
1. Open ScriptManager
- MacOS
- Linux
- Windows
Depending on your system permissions, you may need to be an administrator to open this for the first time. On Mac systems, this can be done by right-clicking the file and selecting ‘Open’ at the top.
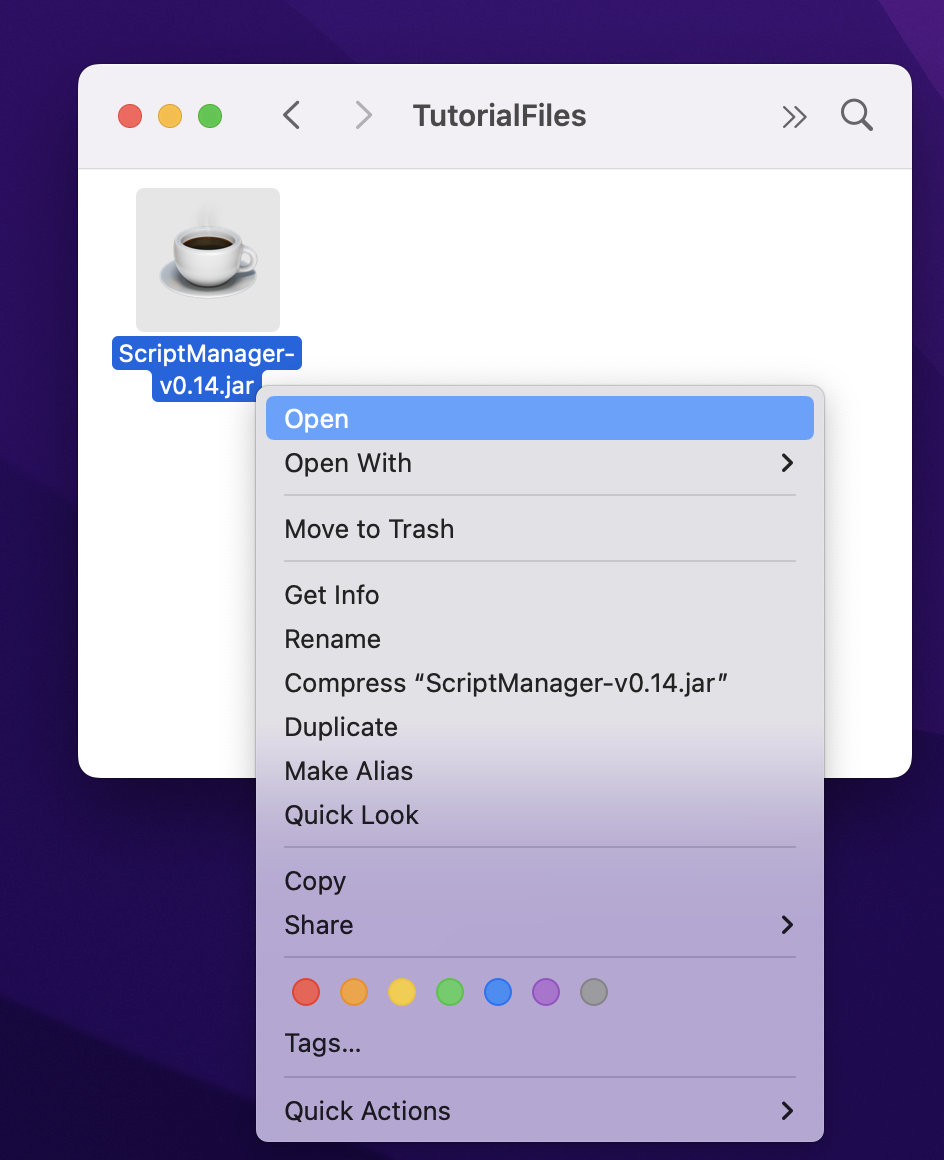
Some MacOS systems may not properly open the JAR by simply double-clicking on the JAR file so you may need to open your Terminal window and execute it from the command line by executing the jar file without any arguments or flags:
java -jar /path/to/ScriptManager.jar
If you're not sure how to type the path to ScriptManager, you can type java -jar (end with space) and then drag ScriptManager from Finder into your Terminal window and then press enter.
Double-click or right-click the ScriptManager JAR file to start the program.

Double-click or right-click the ScriptManager JAR file to start the program.

Once you see the main tool selection window, you're off to the races!
2. Generate BAI index file
A BAI index file is required for each BAM file of interest (i.e., the tag occupancy data you want to plot). This file allows for rapid access of the sorted and aligned sequence reads (BAM file).
SAM/BAM standard is to keep BAI file in same directory as BAM file with the ScriptManager-generated filename.
Reb1_YEP_12141.bam
Reb1_YEP_12141.bam.bai # Need to generate this file to proceed.
2.1. Navigate to BAM Manipulation ➡️ BAM-BAI Indexer

2.2. Generate BAI index files for each BAM file of interest by loading your BAM file and clicking "Index."
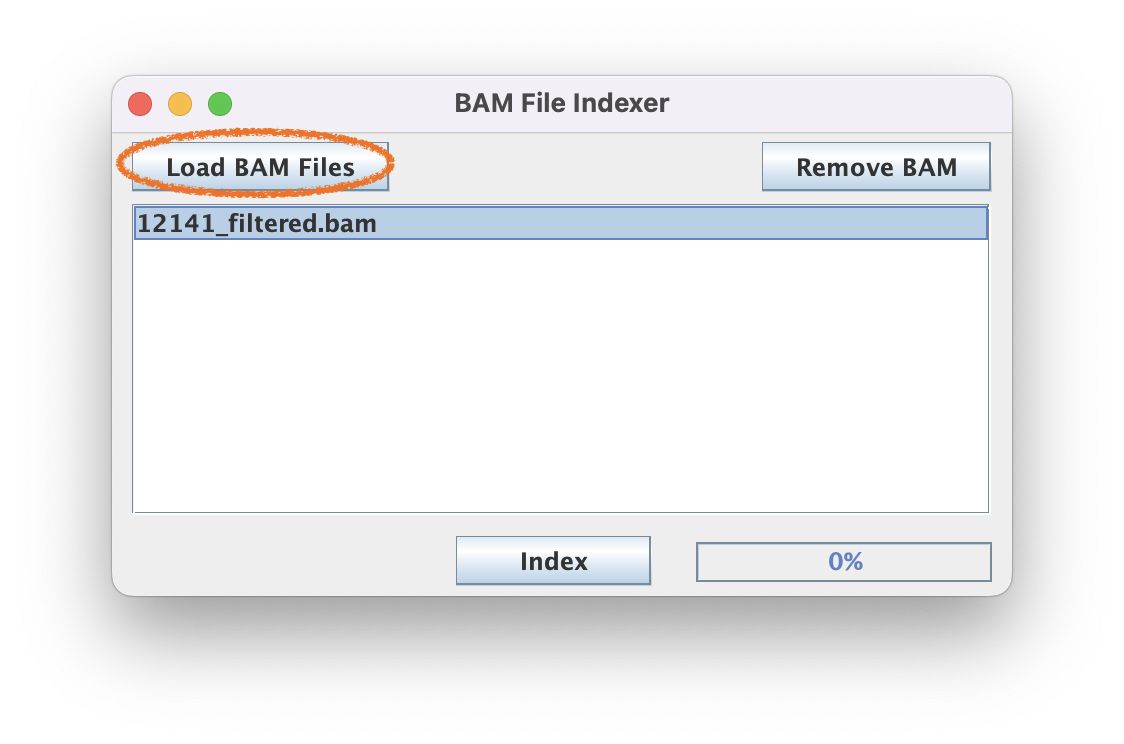
The speed of this step scales with the size of the BAM file. Generally this step 30 sec for a 100 MB BAM file but may take 1-2 min for a multi-GB BAM file.
3. Resize the Reb1 motif-aligned BED file
The BED file is the set of reference coordinates that your heatmap and composite plots will be aligned to, but you’ll need to specify how far upstream and downstream you want your data to be plotted; i.e., “Size of Expansion (bp). If you bed file is defined by more than a 1 bp interval AND you want to add to limits of that interval, then select “Add to Border”).
3.1. Navigate to Coordinate File Manipulation ➡️ Expand BED File

3.2. For this tutorial, use the 250bp expansion and select "Expand from Center".
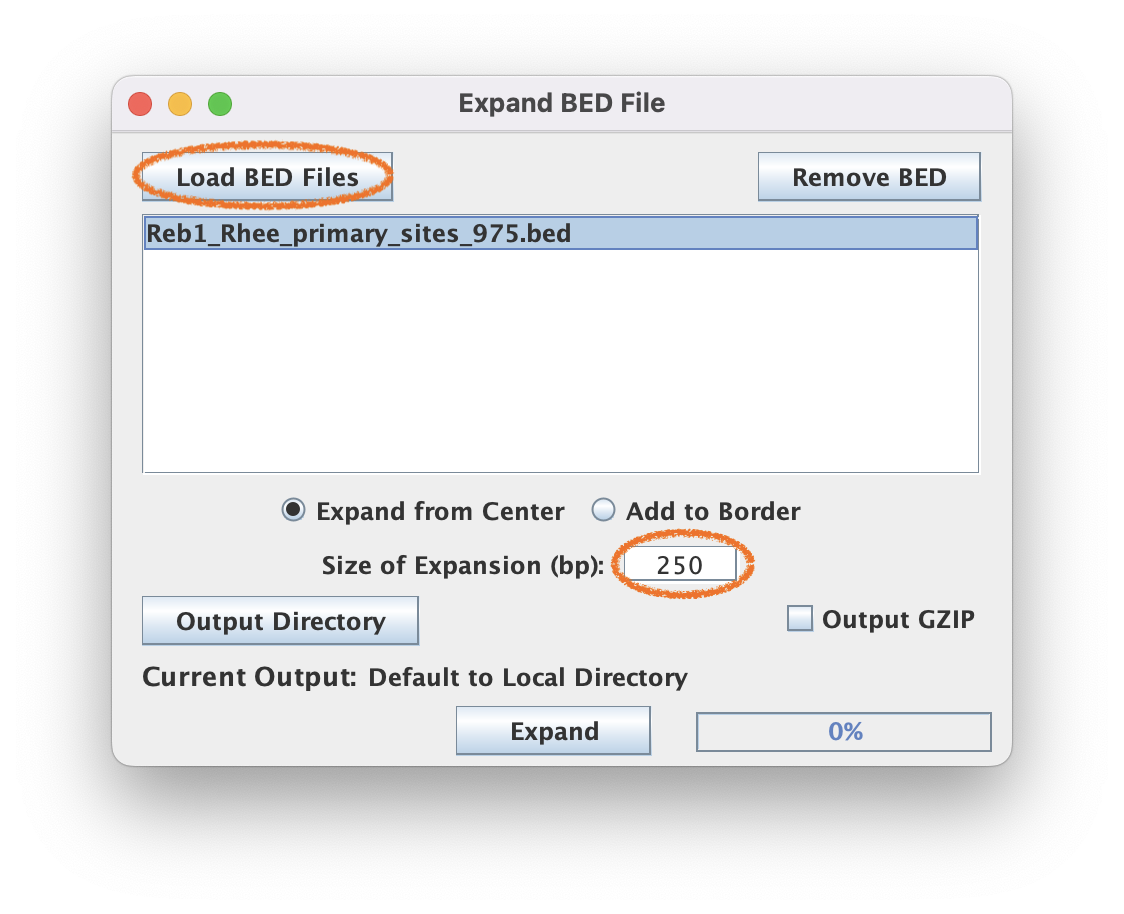

BED file coordinates often need to be resized for more informative tag pileups. For Reb1 (yeast), 250-500 bp windows are generally sufficient. Mammalian samples may require larger windows (500-2000 bp) based on the amount of indirect-crosslinking
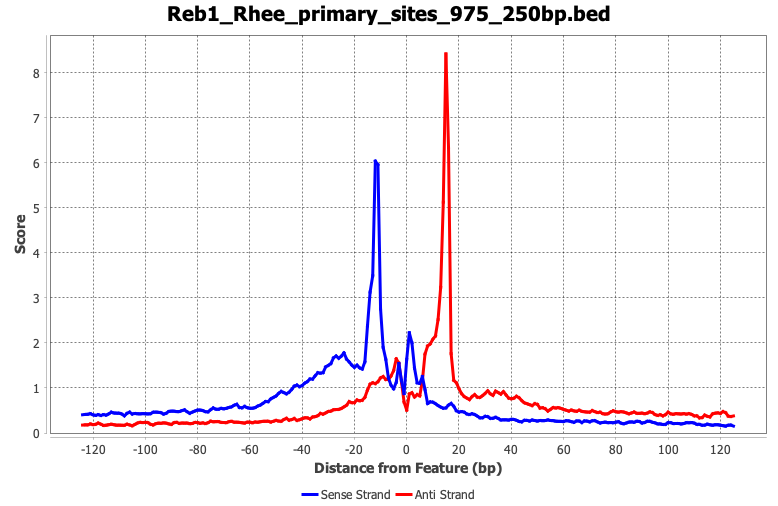
4. Generate the tag pileup
Use TagPileup to pileup the BAM data within a set of BED coordinate windows to generate the composite plot and the matrix(CDT) files that will be used to generate the heatmaps.
4.1. Navigate to Sequence Read Analysis ➡️ Tag Pileup

4.2. Load the BED and BAM files
4.3. Select output directory & make sure "Output Matrix", "CDT", and "Output GZIP" are selected
Bioinformatic projects should be organized in a uniform and consistent manner as described below
Paper on how to organize bioinformatics projects (Noble 2009)
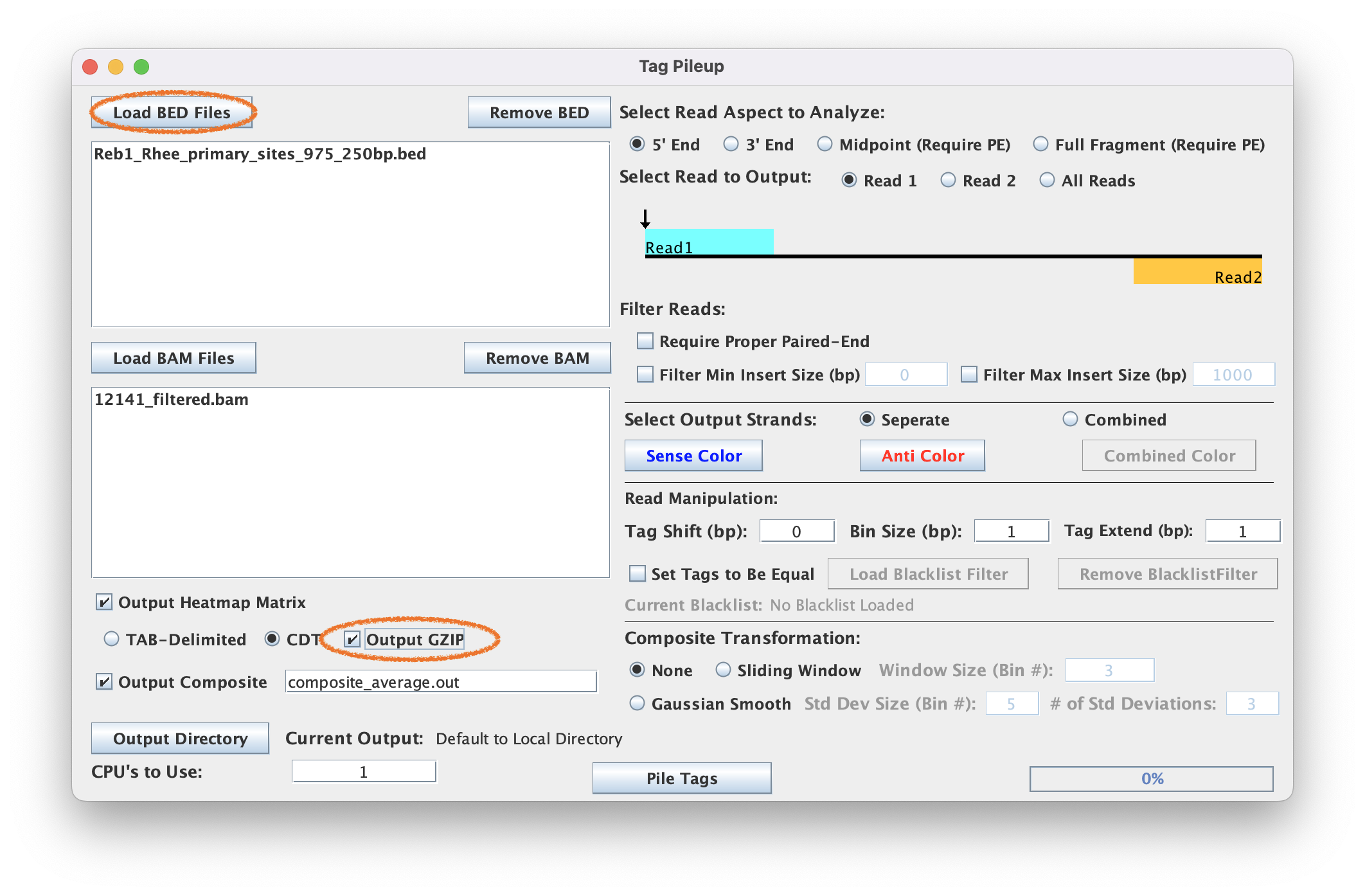
4.4. When ready, select ‘Pile Tags’ to execute
The default parameters Tag Pileup is set to expect is a sequence-specific strand separated ChIP-exo dataset. Modifications to these parameters are needed for more specific analysis or when using data generated from other assays.
4.5. Save composite results
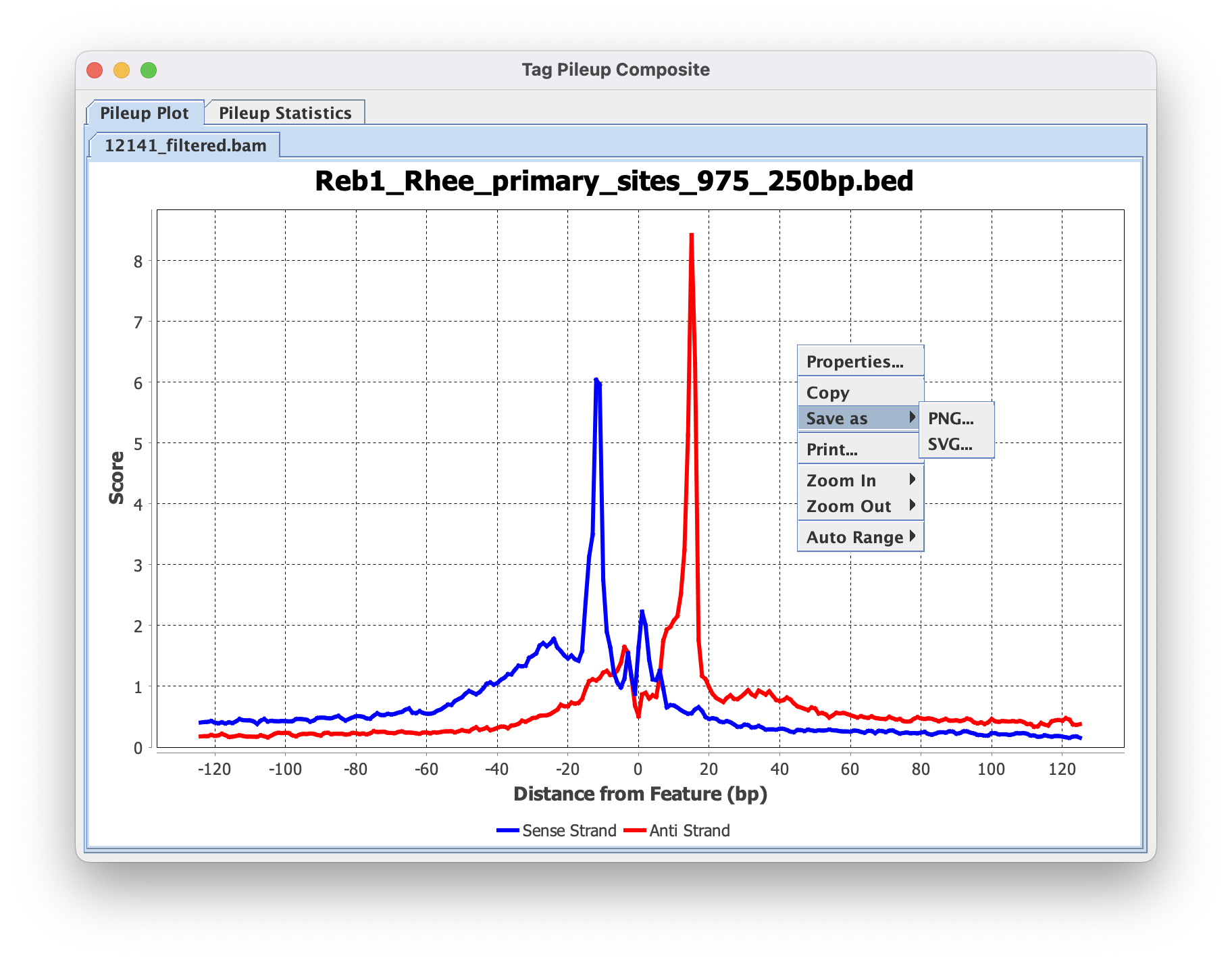
The displayed composite plot can be modified by right-clicking and selecting properties. Things such as axis labels, axis range, and colors can be modified here.
The final image can then be saved by right-clicking and selecting ‘Save as’. PNG is fine for most cases, but SVG is strongly recommended if this composite plot will be used in Adobe Illustrator later.
Besides the composite plot image, ScriptManager has saved the matrix *.CDT files to your Output Directory together with the composite plot values file (If you didn't change the name it would be called composite_average.out). These CDT files will be used as the input for generating heatmaps in the next step.
5. Generate Heatmaps
5.1. Navigate to Figure Generation ➡️ Heat Map.

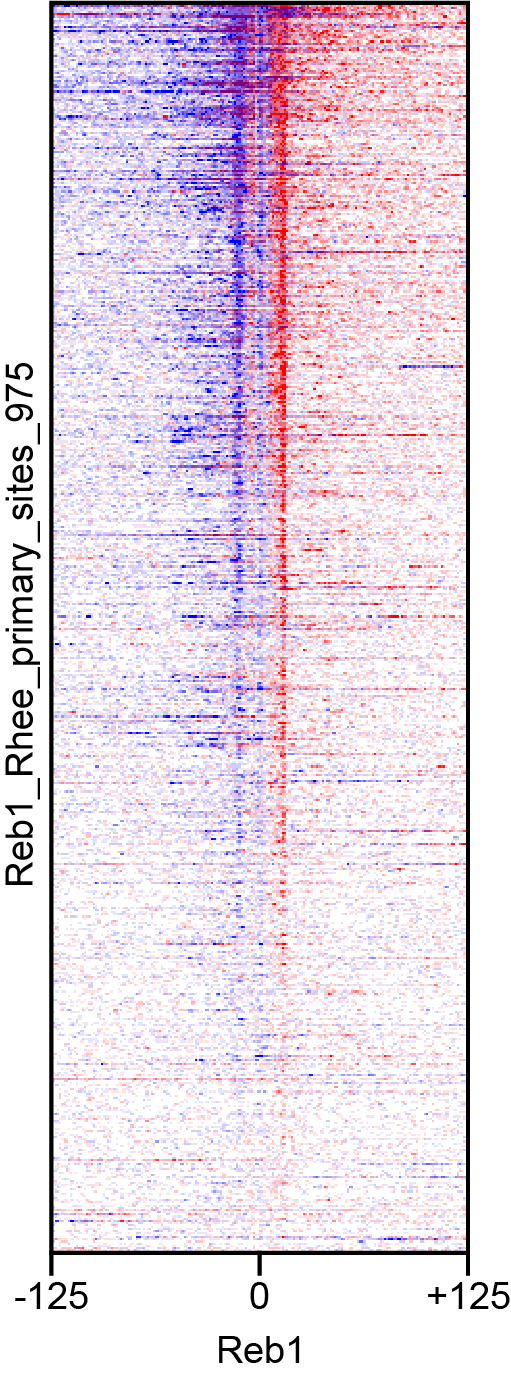
5.2. Heatmap Generator can only generate one color at a time, so ‘Sense’ and ‘Anti’ files should be processed separately. The ChIP-exo standard for strand colors is ‘Sense’ == blue and ‘Anti’ == red.
Start by generating the 'Sense' heatmap first.
5.3. Click "Load Files" and select the _sense.cdt output CDT files from running the TagPileup step.
5.4. Select "blue" for the color (per the lab standard)
5.5. The heatmaps for this dataset show the best contrast when using the default "Percentile Threshold" value (.95 or 95%). Otherwise your heatmaps will come out too light or too dark to see the shape.
5.6. Click the "Output Heatmap" checkbox. The Heatmap generator does not save the produced PNG by default.
5.7. Click "Generate" to save your Sense PNG heatmap!


"Percentile Threshold" is a useful for looking at the shape of ChIP-exo binding patterns while "Absolute Threshold" is useful for setting a shared contrast threshold across several samples and comparing signal density. For other data, you may need to play around with settings to find the right contrast for you data.
Similarly generate the 'Anti' heatmap.
5.8. Remove the "Sense" file by selecting it to highlight it and then clicking the "Remove Files button"
5.9. Click "Load Files" and select the _anti.cdt output CDT files from running the TagPileup step.
5.10. Select "red" for the color (per the lab standard)
5.11. Since all the other parameters should be the same from you "Sense" run, you shouldn't need to re-select the "Percentile Threshold" or "Output Heatmap" options.
5.12. Click "Generate" to save you Anti PNG heatmap!

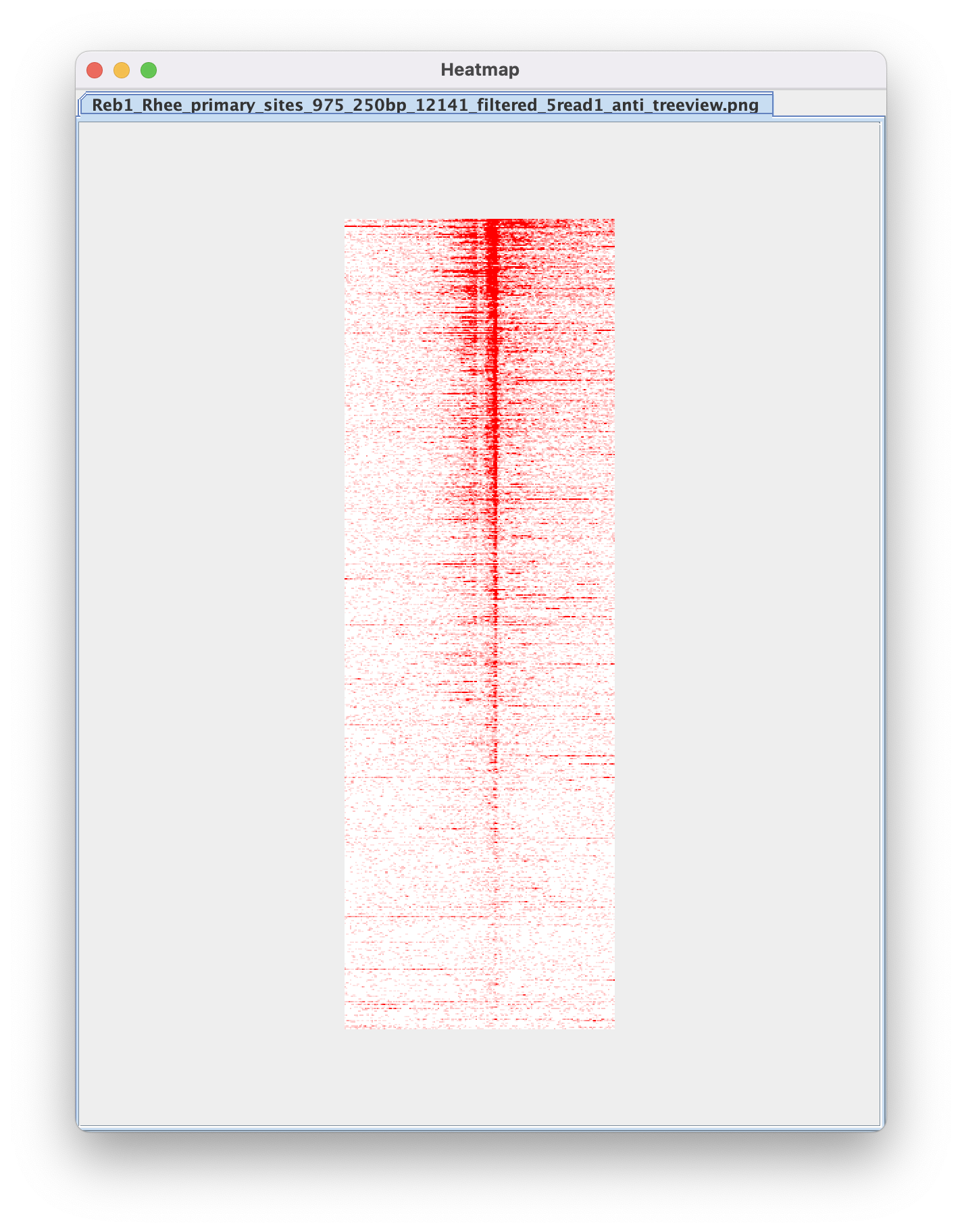
6. Merge strand-separated heatmaps
6.1. Navigate to Figure Generation ➡️ Merge Heatmaps so we can merge our strand-separated heatmaps into a single PNG.
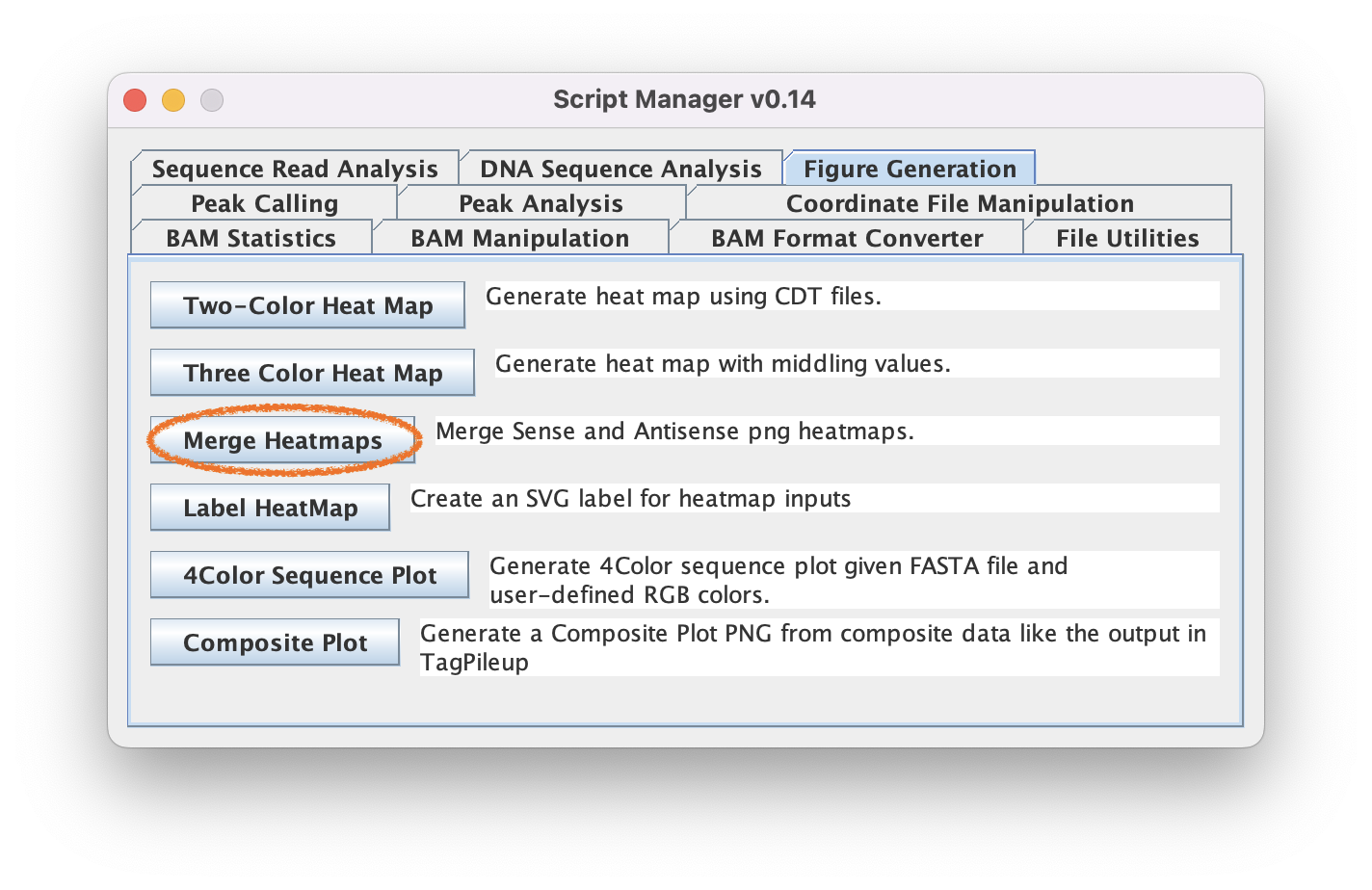
The script will automatically match sense to anti heatmaps using the standardized naming conventions used by ScriptManager
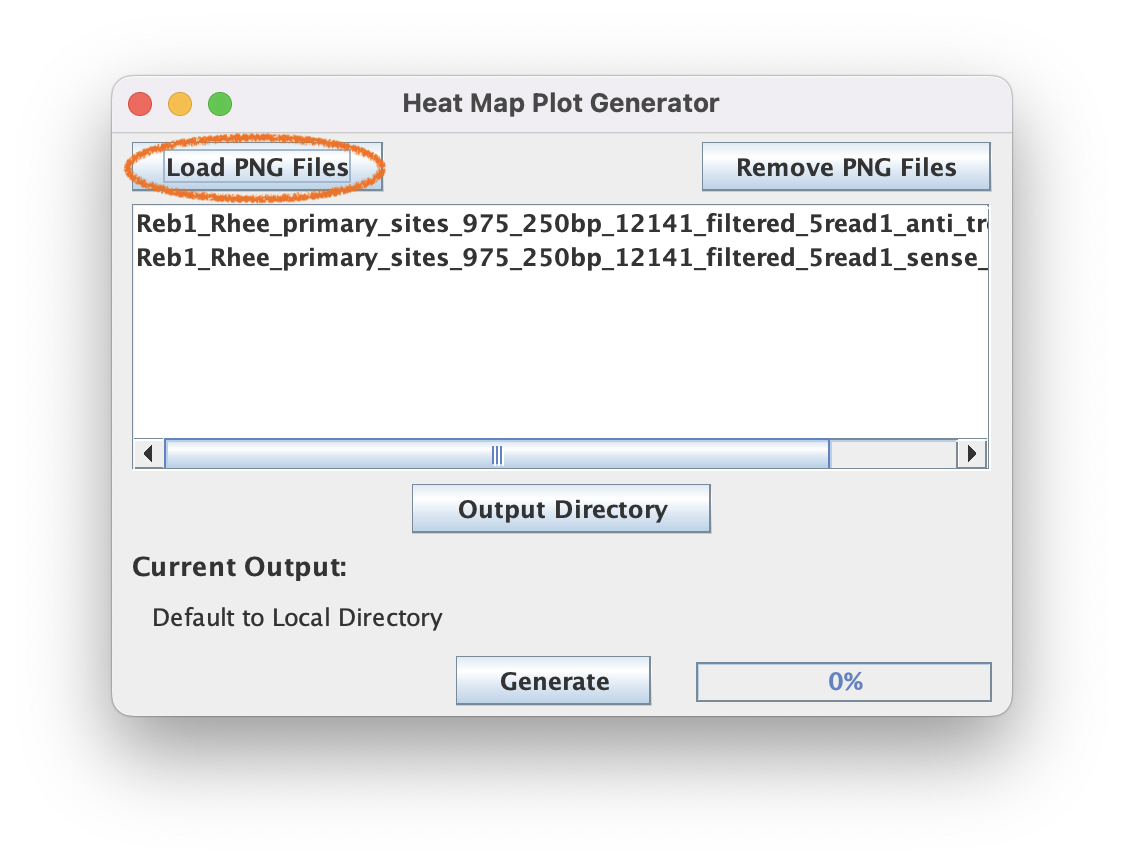
6.2. Click "Load PNG Files" to select the two files output by the HeatMap tool (sense and anti) in the last step
6.3. Click "Generate" to merge the PNG files into the same heatmap

7. Label your merged heatmaps
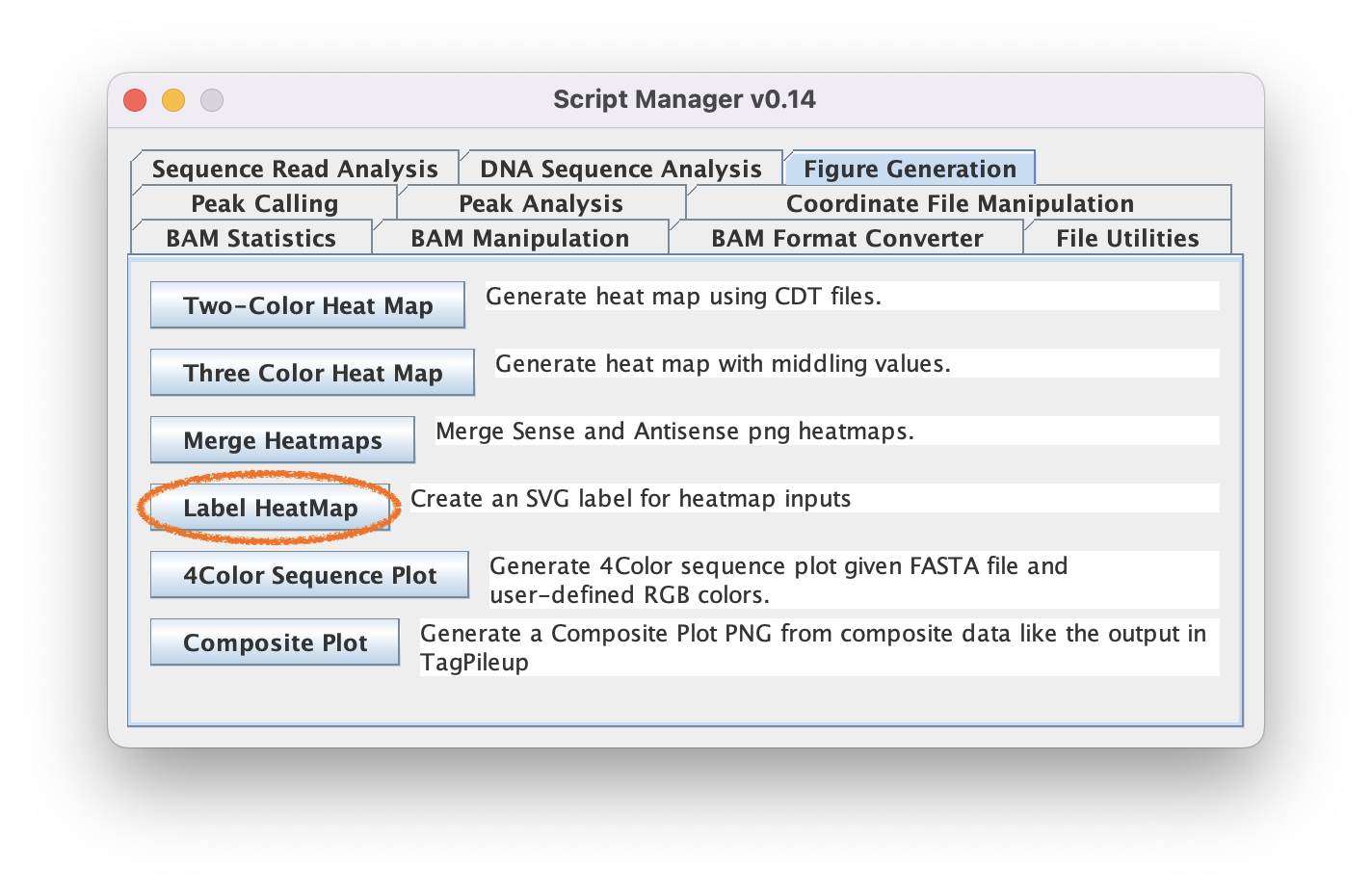
7.1. Navigate to Figure Generation ➡️ Label Heatmaps so we can add axes to the heatmap
7.2. Optionally type text into the fields to annotate your axes (Or leave them empty to just generate an SVG border)
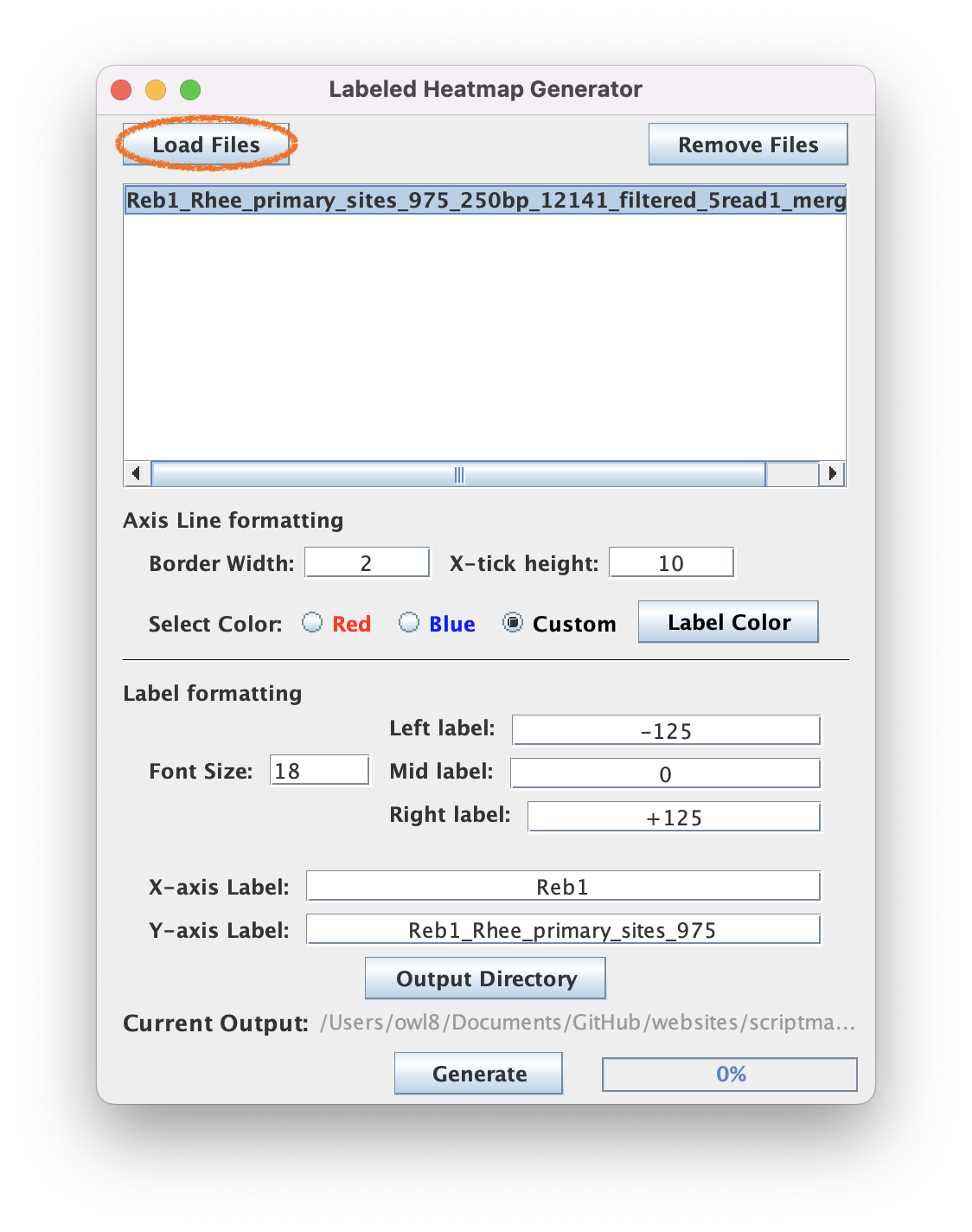
7.3. Click "Generate" to create an SVG file that labels your PNG
Open up your SVG file to see what it looks like! Use PowerPoint, Adobe Illustrator, or wherever you edit your publication figures to make any further edits!
General Comments
Bioinformatic projects should be organized in a uniform and consistent manner so that you can easily find them in the future. Consider the organizational structure described in Noble, 2009.
Command-Line shell script
The following shell commands records the locations for a BED file, a BAM file, and the anticipated OUTPUT basename as environmental variables to derive the corresponding composite plot values and heatmaps. This can serve as a template for you to write out your own workflows as bash scripts that execute command-line style ScriptManager.
SCRIPTMANAGER=/path/to/ScriptManager.jar
BEDFILE=/path/to/Reb1_Rhee_primary_sites_975.bed
BAMFILE=/path/to/12141_filtered.bam
OUTPUT=/path/to/myoutput
samtools index $BAMFILE
java -jar $SCRIPTMANAGER coordinate-manipulation expand-bed -c 250 $BEDFILE -o BED_250bp.bed
java -jar $SCRIPTMANAGER read-analysis tag-pileup $BEDFILE $BAMFILE -o $OUTPUT\_composite.out -M $OUTPUT\_matrix
java -jar $SCRIPTMANAGER figure-generation heatmap -p .95 --blue $OUTPUT\_matrix_sense.cdt -o SENSE.png
java -jar $SCRIPTMANAGER figure-generation heatmap -p .95 --red $OUTPUT\_matrix_anti.cdt -o ANTI.png
java -jar $SCRIPTMANAGER figure-generation merge-heatmap SENSE.png ANTI.png -o $OUTPUT\_heatmap.png
java -jar $SCRIPTMANAGER figure-generation label-heatmap $OUTPUT\_heatmap.png \
-x "Reb1" -y "Reb1_Rhee_primary_sites_975" -l -250 -m 0 -r +250 -o $OUTPUT\_heatmap.svg
rm BED_250bp.bed SENSE.png ANTI.png
# Output files:
# - /path/to/myoutput_composite.out
# - /path/to/myoutput_matrix_sense.cdt
# - /path/to/myoutput_matrix_anti.cdt
# - /path/to/myoutput_heatmap.png