BAM Indexer


Generates BAI Index for input BAM files. Output BAI is in the same directory as input BAM file.
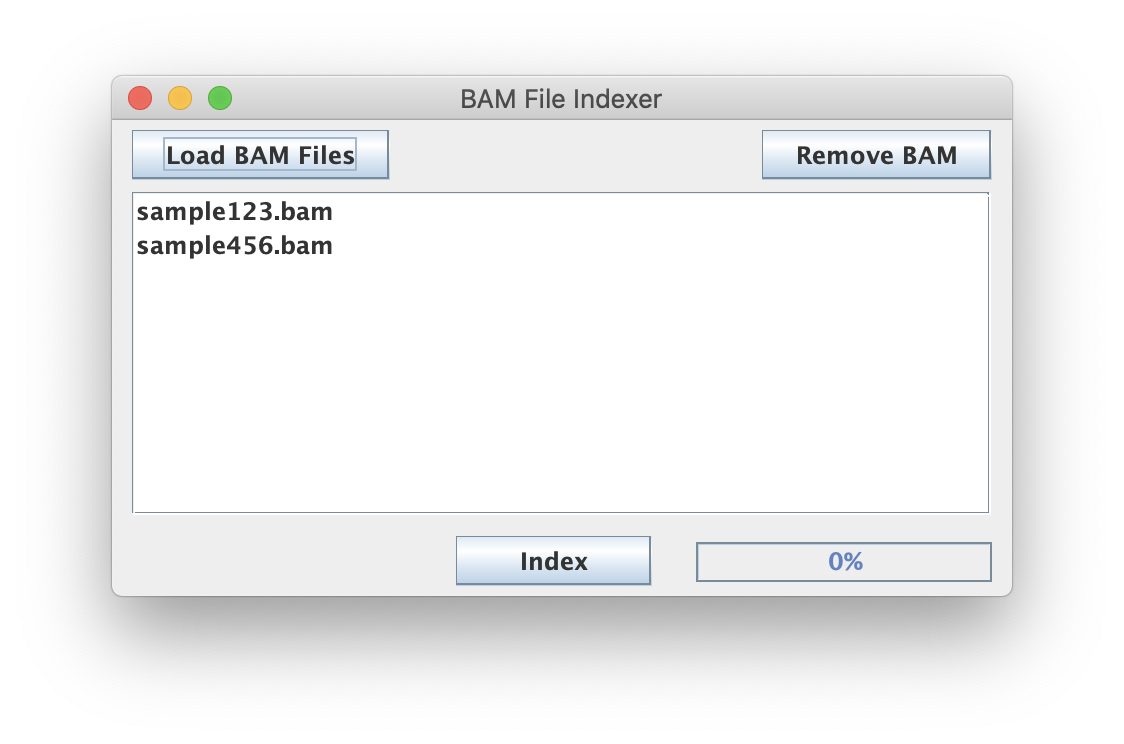
Most tools using BAM inputs (including ScriptManager) require a BAM index file (.bai) file so that the tool can efficiently query the file for alignment records. Read more in the Picard documentation.
ScriptManager's TagPileup, Merge BAM replicates, BAM Correlation, and BAM Format Converter tools (BAM to BED, BAM to GFF, BAM to bedGraph, and BAM to scIDX) are some example tools that require a .bai file.
File inputs (BAM)
Make sure your inputs are properly formatted and use the appropriate .bam extension. This script also supports bulk selection and processing of files.
BAM file MUST be sorted to successfully index.
Output (BAI)
After clicking "Index", ScriptManager will index all of the loaded BAM files and save them with the .bai extension following BAI convention.
It is standard practice to generate and save the index file in the same place with the same name as the .bam file it is indexing so that your bioinformatics tools can find it.
For example, the file /User/bob/mydir/sample123.bam will be indexed to /User/bob/mydir/sample123.bam.bai.
Command Line Interface (Picard and Samtools)
CommandLine tools already exist for this function. This tool only exists as a GUI wrapper in ScriptManager.
Please see the Samtools index tool or the Picard BuildBamIndex tool.