Mark Duplicates (Picard)


Removes or marks duplicate reads in paired-end sequencing given identical 5' read positions. Read more in the Picard documentation.
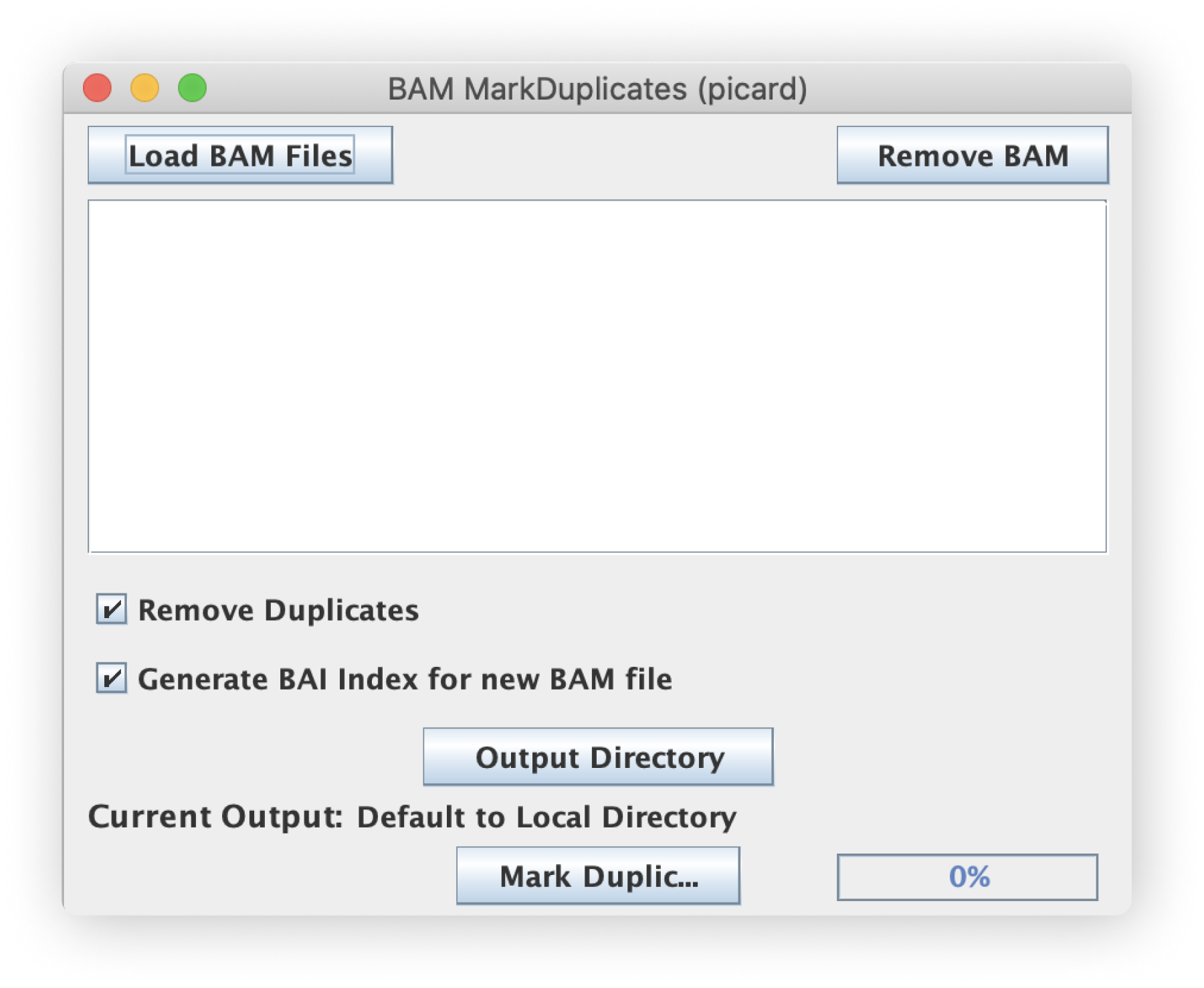
File inputs (BAM)
This script filters BAM-type files so make sure your inputs are properly formatted and use the appropriate .bam extension. The script also supports bulk selection and processing of files.
Output file (BAM & TXT)
The output BAM files are named based on the input filenames. The _dedup.bam suffix is used for each output. For example, for a given XXX.bam input file, a new XXX_dedup.bam file will be written to the user-selected output directory.
The output text files are also named based on the input filenames. The _dedup.metrics suffix is used for each output. For example, for a given XXX.bam input file, a new XXX_dedup.metrics file will be written to the user-selected output directory.
Make sure if you change the output BAM filename that you keep the .bam file extension.
Example XXX_dedup.metrix output (TXT)
Remove Duplicates Option
Each output file can either remove one of the duplicate reads or mark them by changing the SAMFlag value to update the status as "duplicate." This checkbox option determines whether the duplicate reads are kept or just marked.
Generate BAI file (GUI only)
By checking this box, the script will automatically generate a BAI index file for each new filtered BAM file.
Command Line Interface (Picard and Samtools)
CommandLine tools already exist for this function. This tool only exists as a GUI wrapper in ScriptManager.
Please see the Samtools markdup tool or the Picard MarkDuplicates tool for a command line tool that performs this function.