Filter PIP-seq

Filter BAM file by -1 nucleotide. Requires genome FASTA file.
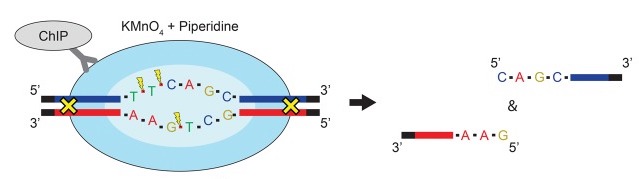
Permanganate (KMnO4) and piperidine treatment of DNA fragments preferentially oxidizes and cleaves off the T (thymine) at the 5' end of single stranded DNA fragment. When analyzing data from sequencing assays like PIP-seq that use this treatment (Lai et al, 2017), a filter step using this tool for reads that align to positions with a 'T' at the -1 position of the 5' end of read 1 can reduce the amount of noise (i.e. DNA fragments not cleaved by piperidine). This tool can potentially clarify signal in the downstream steps of your analysis.
Genome input (FASTA)
Since the alignment files only capture the reference genome sequence at genomic positions covered by a read, the sequence upstream of the 5' end of Read 1 is not necessarily captured by the BAM file format. Thus the reference genome is required to determine the sequence upstream of the 5' end of read 1 (the basis for this filtering script).
Make sure that the genome build used for the genome input matches the genome aligned to for the BAM formatted files. If you aren't sure, compare the chromosome lengths in the genomic FASTA index file (FAI) against each BAM file header (samtools view -H myfile.bam).
File inputs (BAM)
This script filters BAM-type files so make sure your inputs are properly formatted and use the appropriate .bam extension. The script also supports bulk selection and processing of files.
Output
The output file for this script is a filtered set of alignments in BAM format for each input BAM file. The _PSfilter.bam suffix is used for each output. For example, for a given XXX.bam input file, XXX_PSfilter.bam will be output to the user-selected output directory.
Upstream sequence
The sequence upstream of the 5' end of read 1 to check for and filter by. If the sequence in the reference genome upstream of the 5'end of read 1 matches this sequence, the read-pair information is written to the output BAM file.
For classic PIP-seq datasets the default "T" sequence should be used.
This tool supports different sequences in the event of yet unknown future biochemical assay or analysis requires this filtering based on a different sequence. For example, a user investigating and comparing the rates of permanganate oxidation and piperdine cleavage at other nucelotides might compare BAM files filtered by other upstream sequences such as "C" which is known to be cleaved under such treatment (just not as frequently as at "T").
Generate BAI file (GUI only)
By checking this box, the script will automatically generate a BAI index file for each new output BAM file.
Command Line Interface
Usage:
java -jar ScriptManager.jar bam-manipulation filter-pip-seq [-hV] [-f=<filterString>]
[-o=<output>] <genomeFASTA> <bamFile>
Output Options
| Option | Description |
|---|---|
-o, --output=<output> | specify output file (default=<bamFileNoExt>_PSfilter.bam) |
-f, --filter=<filterString> | filter by upstream sequence, works only for single-nucleotide A,T,C, or G. (default seq='T') |