Merge BAM


Merges Multiple BAM files into single BAM file. Sorting is performed automatically. RAM intensive process. If program freezes, increase JAVA heap size.
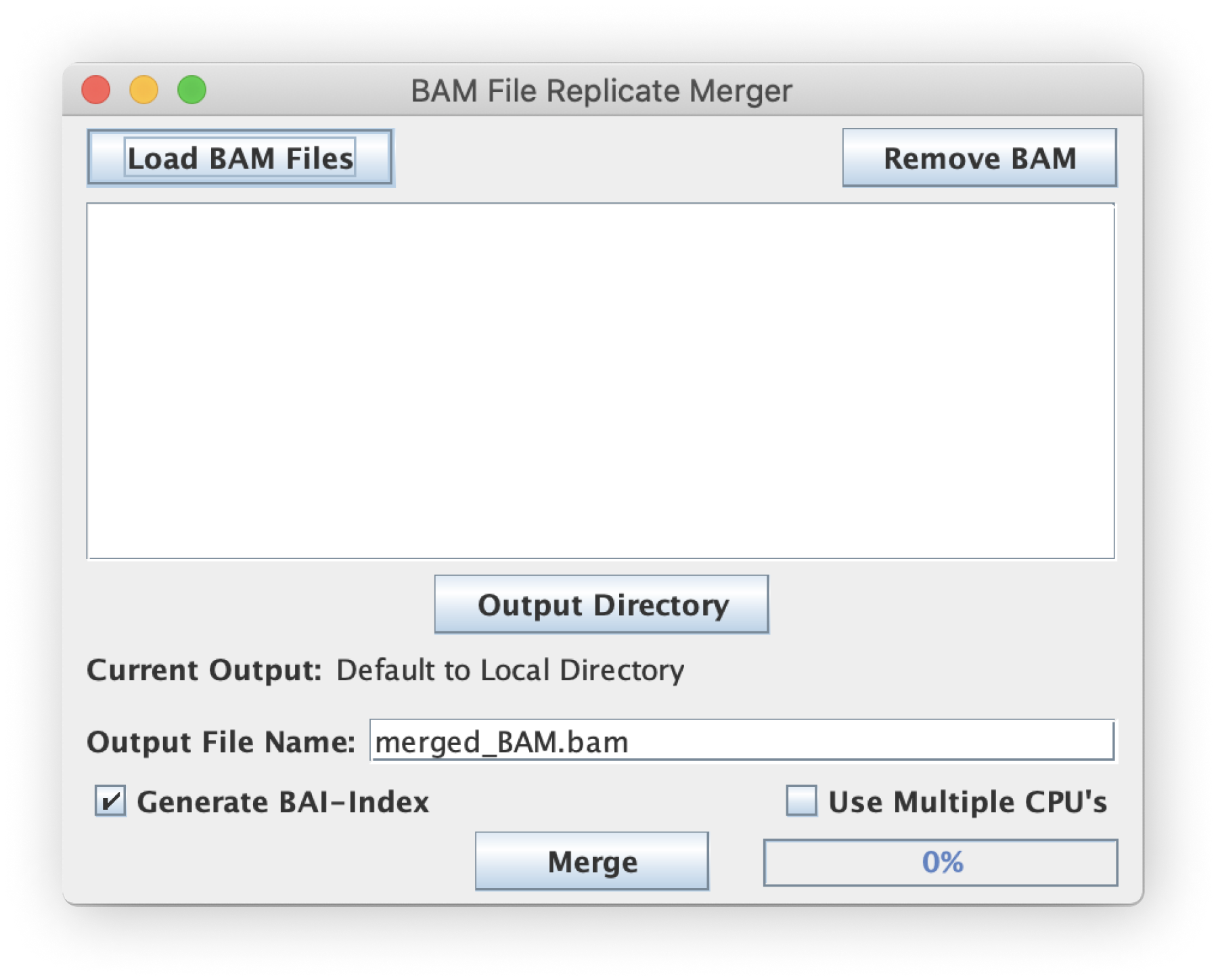
This is frequently used for replicate merging. All input files loaded will be merged to a single BAM file.
File inputs (BAM list)
Make sure your inputs are properly formatted and use the appropriate .bam extension. This script also supports bulk selection and processing of files.
Output File Name (BAM)
The output merged BAM will be named according to the user-customizable text field that defaults to merged_BAM.bam in the user-selected "Output Directory".
Make sure if you change the output BAM filename that you keep the .bam file extension.
Use multiple CPUs
User may speed up the merging by checking this box to allow threading for parallelization of the merge and sort algorithms.
Generate BAI file (GUI only)
By checking this box, the script will automatically generate a BAI index file for each new filtered BAM file.
Command Line Interface (Picard and Samtools)
CommandLine tools already exist for this function. This tool only exists as a GUI wrapper in ScriptManager.
Please see the Samtools merge tool or the Picard MergeBamAlignment tool for a command line tool that performs this function.