BAM to GFF

Convert BAM file to GFF file
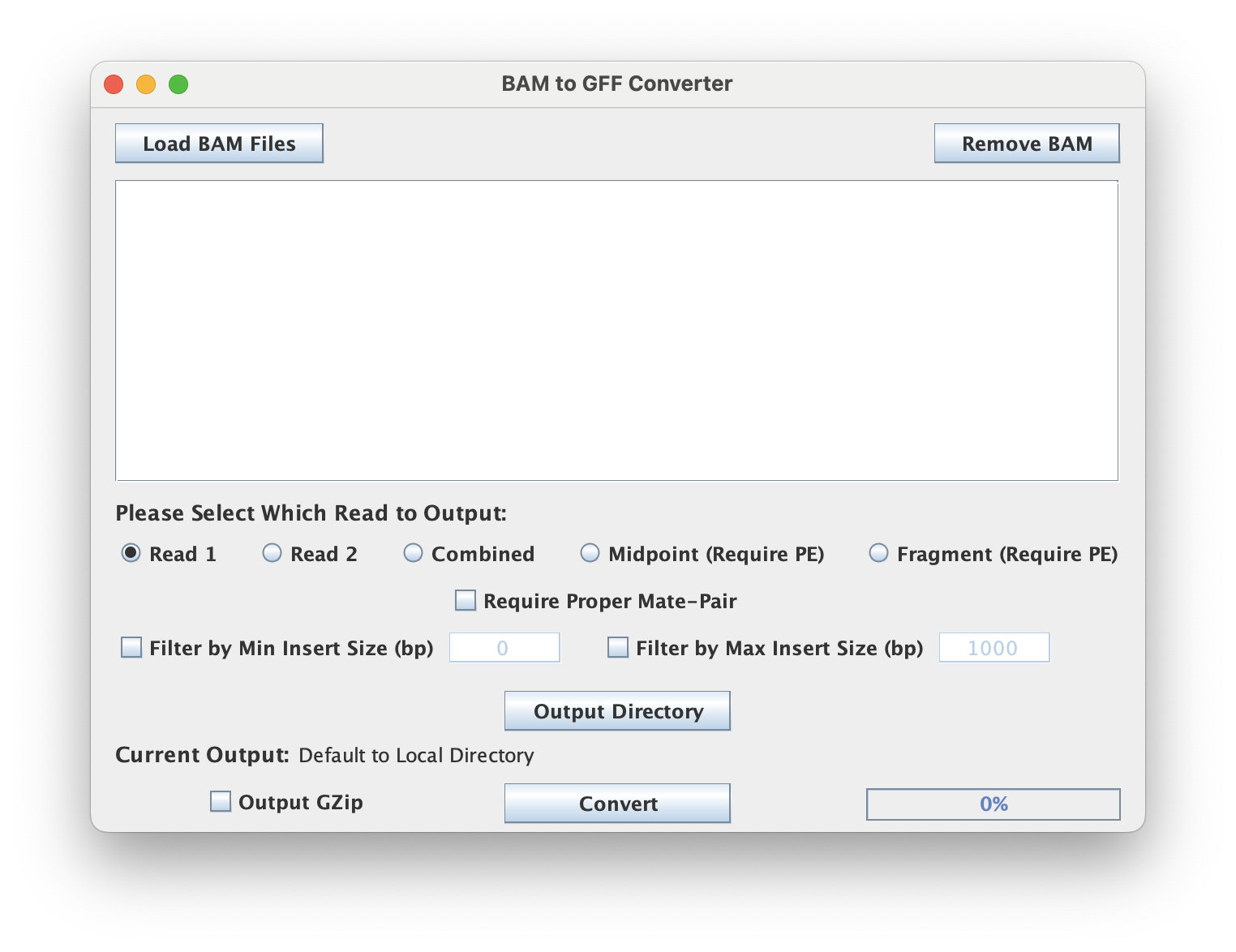
Read Options
This tool has multiple read options to output.
- Read 1: output Read 1
- Read 2: output Read 2
- Combined: output combined reads
- Midpoint: output the midpoint between reads
- Fragment: output the full fragment of two reads
Note: The Midpoint and Fragment options requires proper mate-pair reading.
Command Line Interface
Usage:
java -jar ScriptManager.jar bam-format-converter bam-to-gff [-1 | -2 | -a | -m | -f]
[-hpsV] [-n=<MIN_INSERT>] [-o=<output>] [-x=<MAX_INSERT>] <bamFile>
Positional Input
This tool takes a single BAM file for input. As with other tools, this tool requires the BAM file be indexed.
Output Options
| Option | Description |
|---|---|
-o, --output=<output> | specify output directory (name will be same as original with .gff ext) |
-s, --stdout | stream output file to STDOUT (cannot be used with -o flag) |
-z, --gzip | gzip output (default=false) |
Filter Options
These filter options are shared across all the BAM Format Converter tools.
| Option | Description |
|---|---|
-p, --mate-pair | require proper mate pair (default not required) |
-n, --min-insert=<MIN_INSERT> | filter by min insert size in bp |
-x, --max-insert=<MAX_INSERT> | filter by max insert size in bp |
Read Options
| Option | Description |
|---|---|
-1, --read1 | output read 1 (default) |
-2, --read2 | output read 2 |
-a, --all-reads | output combined |
-m, --midpoint | output midpoint (require PE) |
-f, --fragment | output fragment (requires PE) |