BAM Correlation

Genome-Genome correlations for replicate comparisons given multiple sorted and indexed (BAI) BAM files.
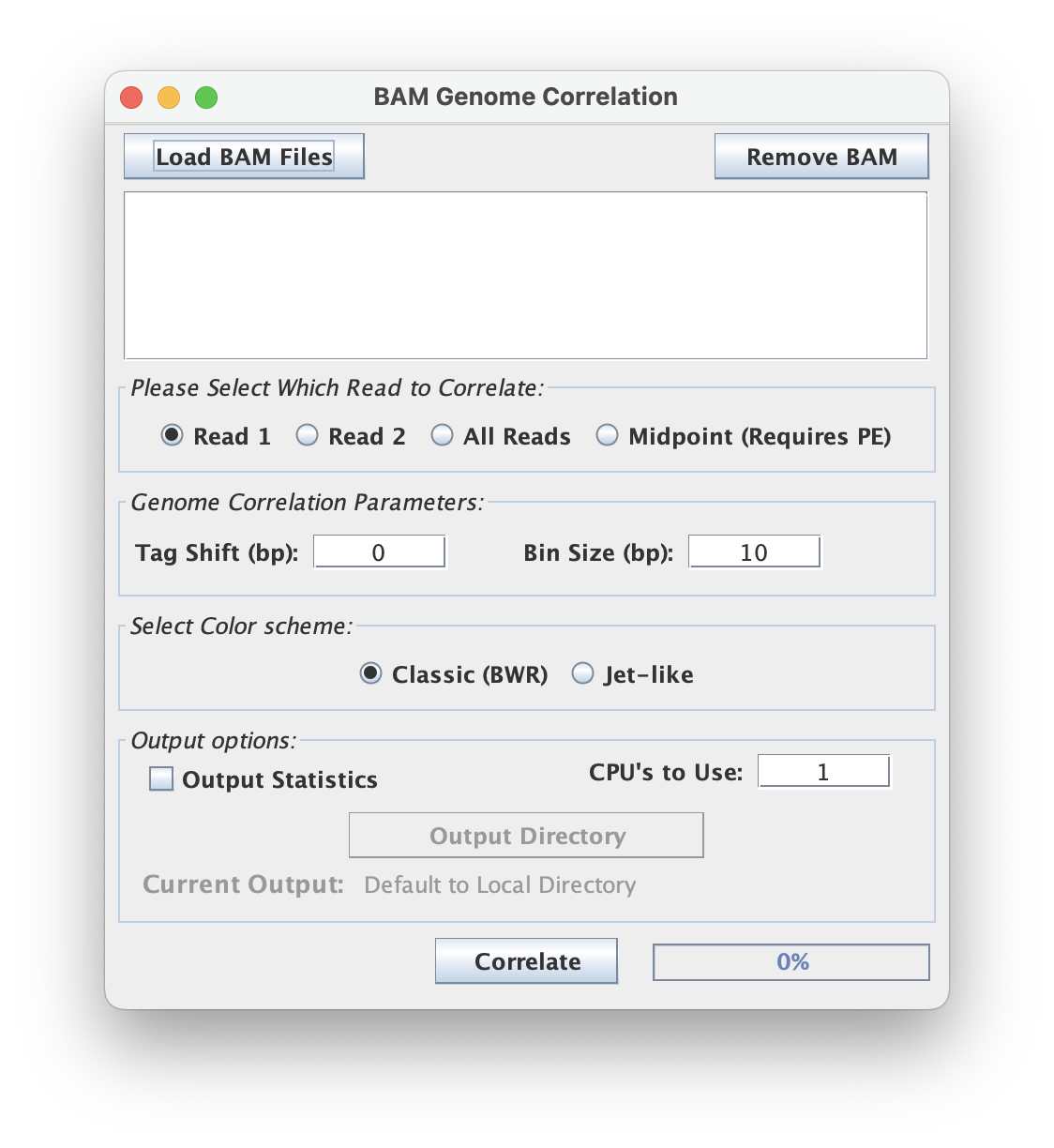
Read Options
This tool has multiple read options to output.
- Read 1: output Read 1
- Read 2: output Read 2
- All Reads: output both reads combined
- Midpoint (Requires PE): output the midpoint between reads
Command Line Interface
Compare a list of BAM files to get a matrix of correlations between them. Outputs both a text file of matrix correlation scores and a heatmap PNG.
Usage:
java -jar ScriptManager.jar bam-statistics bam-correlation
[-1 | -2 | -a | -m] [-fhV] [-b=<binSize>] [--cpu=<cpu>]
[-o=<outputBasename>] [-t=<tagshift>] [<inputFiles>...]
Input Options
| Option | Description |
|---|---|
<inputFiles>... | The BAM file(s) whose statistics we want. |
-f, --files | Input file list of BAM filepaths to correlate (formatted so each path is on its own line) |
Since this tool process a bunch of files together, there are two ways of feeding input files:
(1) You can list them out in the command line tool,
java -jar ScriptManager.jar bam-statistics bam-correlation
bamFile1 bamFile2 ... bamFileX <OPTIONS>
(2) or you can write all the paths for all your files in a single file and pass that as the input using the -f flag
java -jar ScriptManager.jar bam-statistics bam-correlation inputFile -f <OPTIONS>
...where inputFile is listed out line by line:
/path/to/bamFile1
/path/to/bamFile2
...
/path/to/bamFileX
Note that absolute file paths are easier to work with. For relative paths, you\'ll have to check that they are built with respect to the ScriptManager directory.
Output Options
| Option | Description |
|---|---|
-o, --output=<outputBasename> | Specify output file, default is "correlation_matrix" or the input filename if -f flag used |
| Option | Description |
|---|---|
-t, --tag-shift=<tagshift> | tag shift in bp (default 0) |
-b, --bin-size=<binSize> | bin size in bp (default 10) |
--cpu=<cpu> | CPUs to use (default 1) |
Read Options
| Option | Description |
|---|---|
-1, --read1 | output read 1 (default) |
-2, --read2 | output read 2 |
-a, --all-reads | output combined |
-m, --midpoint | output midpoint (require PE) |