Paired-End Statistics

Generates Insert-size Histogram statistic (GEO requirement) and outputs BAM Header including alignment statistics and parameters given a sorted and indexed (BAI) paired-end BAM File.
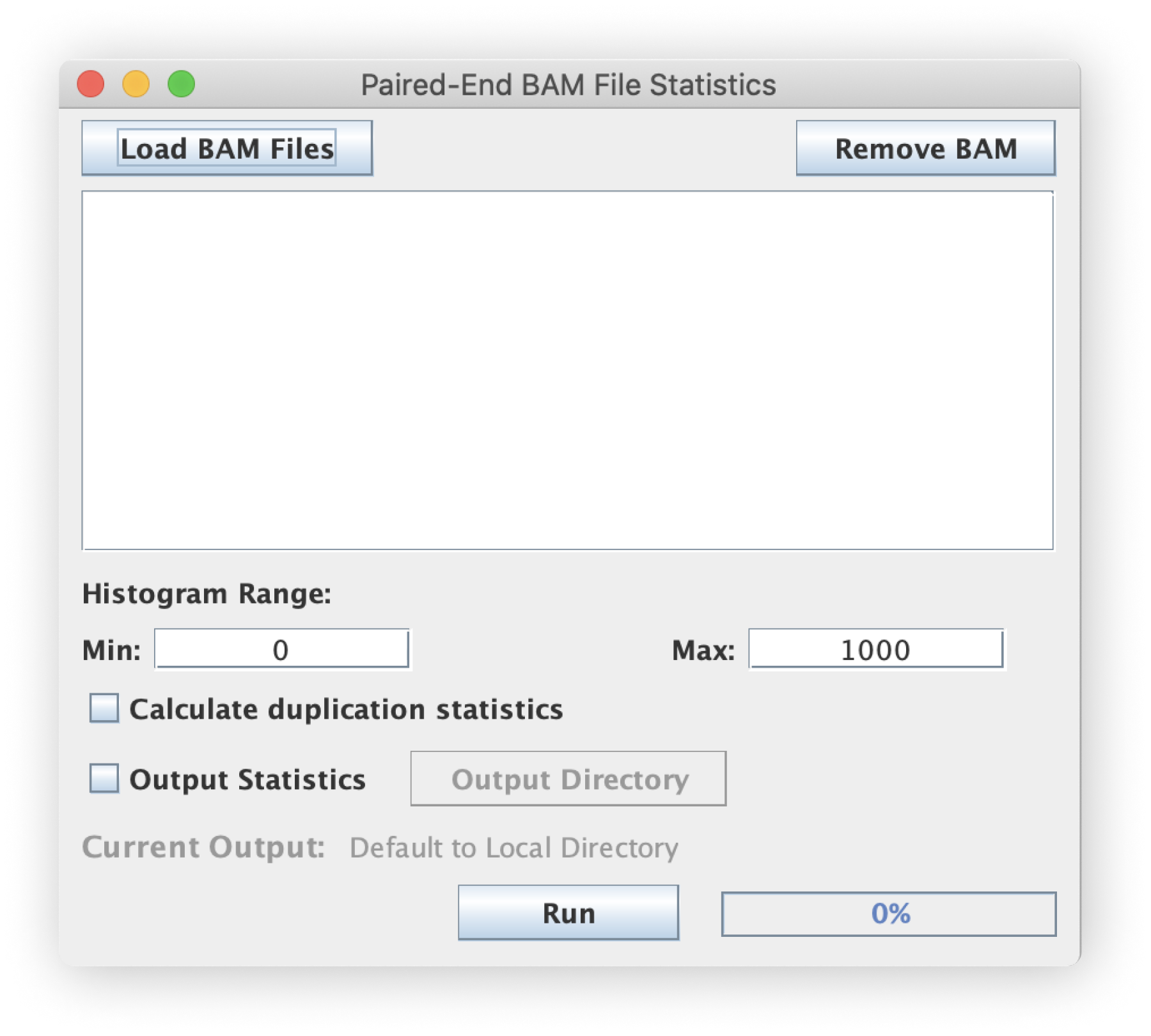
This tool processes each input BAM file by calculating and tallying the insert-size of every single read pair.
Duplication Statistics
The user can determine the duplicate rate vs. number of duplicate molecules by checking the box "Calculate duplication statistics."
Command Line Interface
Usage:
java -jar ScriptManager.jar bam-statistics pe-stat <bamFile> [-dhsV] [-n=<MIN_INSERT>]
[-o=<outputBasename>] [-x=<MAX_INSERT>]
Positional Input
This tool takes a single BAM file for input. As with other tools, this tool requires the BAM file be indexed.
Output Options
| Option | Description |
|---|---|
-o, --output=<outputBasename> | specify output basename, default is the BAM input filename without extension |
-s, --summary | write summary of insert histogram by chromosome (default false) |
-d, --duplication-stats | calculate duplication statistics if this flag is used (default false) |
Filter Options
| Option | Description |
|---|---|
-n, --min=<MIN_INSERT> | histogram range minimum (0 default) |
-x, --max=<MAX_INSERT> | histogram range maximum (1000 default) |