BAM Cross Correlation (ArchTEx)
Calculate optimal tag shift based on ArchTEx implementation from Lai et al, 2012 (PMID:22302569).
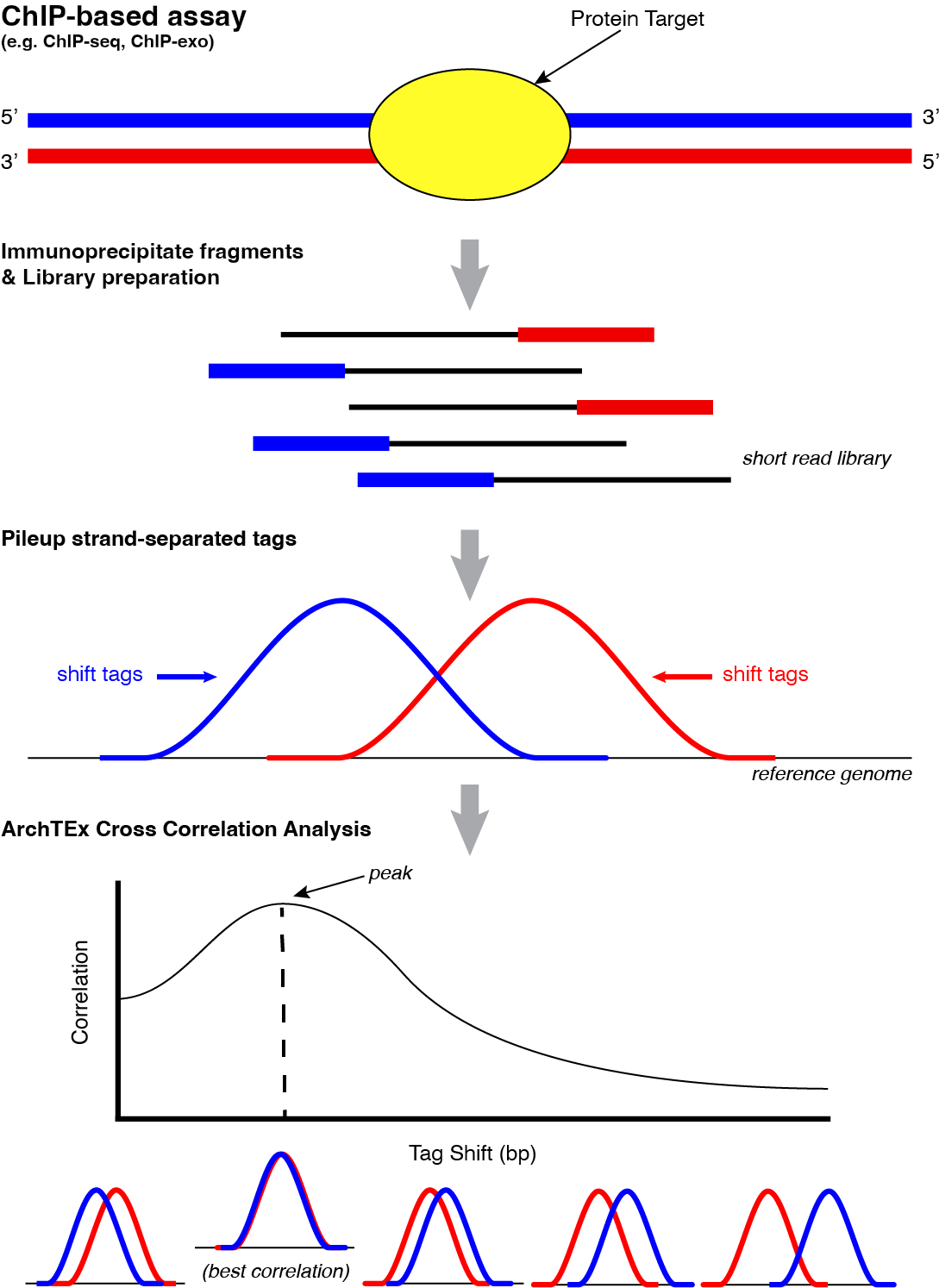
This tool is typically used for empirically determining a tag shift value for combining forward and reverse strand occupancies in tools such as Tag Pileup. In many chromatin immunoprecipitation (ChIP)-based approaches, there is a slight offset of strand-specific read occupancies as shown above and this offset is different across experiments due to differences in the fragmentation method, the length of DNA that the protein target binds, secondary crosslink patterning, and a variety of other factors. To account for this variable offset, this computational approach to determining tag shift was developed. The correlation for every tag shift from 0 to 1000bp is calculated to determine the shift with the best correlation within this range.
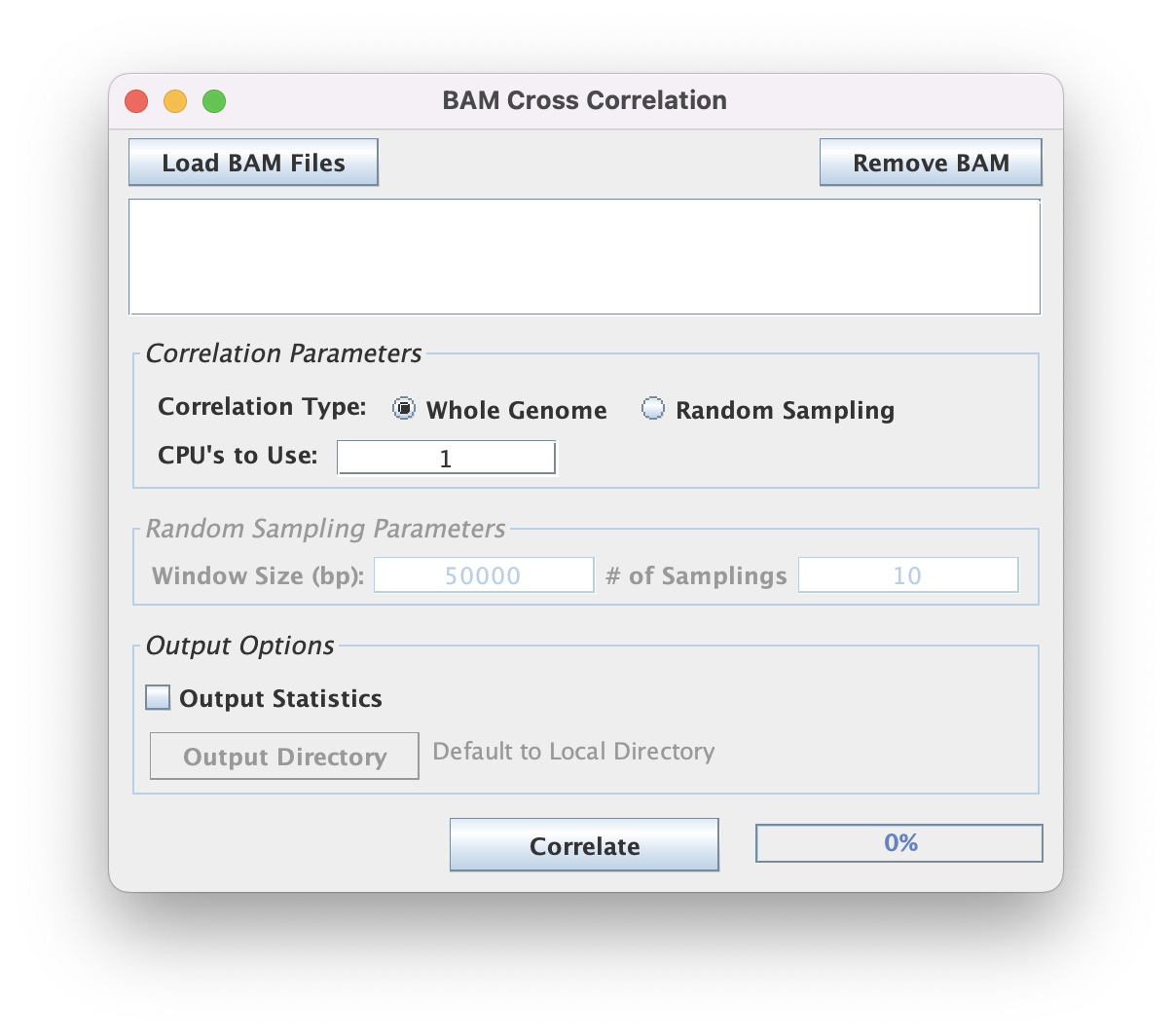
Input BAM files
The graphical interface restricts file selection by the .bam file extension. This tool supports batch processing of files.
Correlation Strategies
You may choose from two strategies for calculating the correlation:
- Whole Genome (recommended) correlation will calculate the correlation of tags across the entire genome (chr sizes inferred from BAM header).
- Random sampling correlation will sample some user-specified number of sites from each chromosome across a user-specified sized window.
Output Files
You may optionally select to write the correlations scores and peak position output to a text file by checking the "Output Statistics" checkbox.
The output window will display progress through the file (printing chromosome currently being analyzed) as well as final Tag Shift (x-axis) ➡️ Correlation Values (y-axis) under the "C-C Data" tab. The "C-C Plots" tab will display the same values as a line plot.
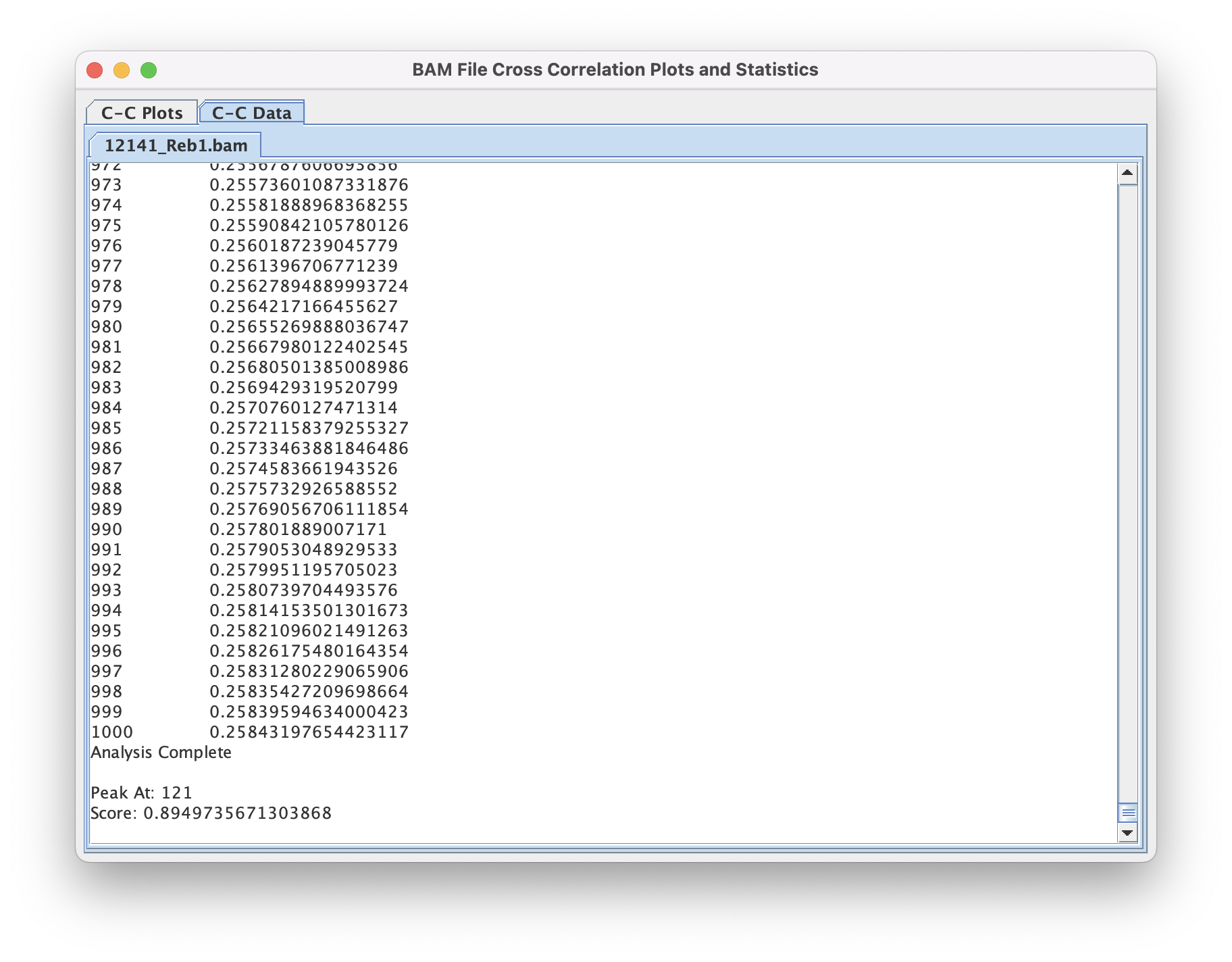
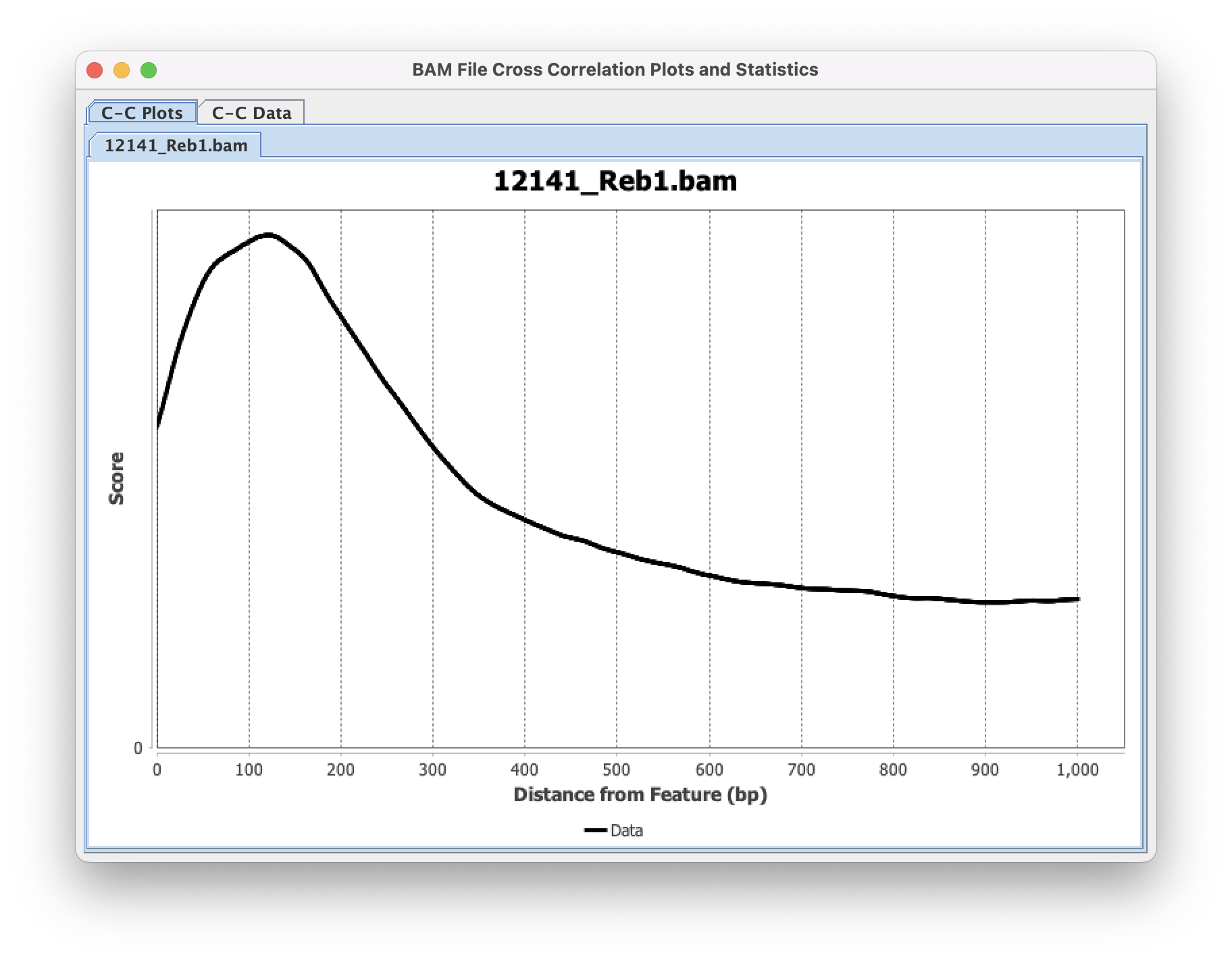
Command Line Interface
Usage:
java -jar ScriptManager.jar bam-statistics cross-corr [-g | -r]
[-w=<windowSize> | -i=<iterations>] [-hV] [-o=<outputBasename>]
[-t=<cpu>] <bamFile>
Positional Input
This tool takes a single BAM file for input. As with other tools, this tool requires the BAM file be indexed.
Output Option
| Option | Description |
|---|---|
-o, --output=<outputBasename> | specify output file basename for correlation scores |
Correlation Strategy Options
Select no more than one strategy.
| Option | Description |
|---|---|
-r, --random | Use the random sampling correlation method (default) |
-g, --genome | Use the full genome correlation method |
If random sampling is selected (-r), you may select more options to specify how sampling is performed. These are otherwise ignored.
| Option | Description |
|---|---|
-w, --window=<windowSize> | set window frame size for each extraction (default=50kb) |
-i, --iterations=<iterations> | set number of random iterations per chromosome (default=10) |
Other Options
| Option | Description |
|---|---|
-t, --cpu=<cpu> | set number of threads for performance tuning (default=1) |
Original code: https://github.com/WilliamKMLai/ArchTEx