Making Genome Tracks Tutorial
How to turn your BAM files into genome track files that can be viewed in a genome browser
Goal: This tutorial provides a guide to transforming and filtering your ChIP-exo alignment data into files that can be uploaded by a genome browser.
❗ BigWig Warning ❗ Be careful about performing analysis steps using BigWig files instead of BAM files. BigWigs are excellent for visualizing data pileups in a genome browser but if you are processing data, we recommend you process data directly from BAM formats. From a information content and reproducibility standpoint, BAM files have notable advantages over BigWigs. Read more about some of these advantages
This tutorial is based on the processing script from Mittal et al, 2022 to turn BAM files into BigWig tracks with base-pair resolution for high resolution assays.
There will be a couple steps that need to be performed on the command line using UCSC binaries because we currently don't have a parser for BigWig files (in the works but not ready yet). In the meantime, here are the step-by-step instructions for using as much of ScriptManager's GUI as possible to generate the BigWig track.
Download ScriptManager (v0.14):
The current version of ScriptManager is available for download here. Make sure you have Java installed.
The file ScriptManager-v0.14.jar should be placed someplace locally accessible. For example on Mac OS on the Desktop (Permissions will need to be accepted) or someplace in your home directory (i.e. Macintosh HD/Users/userID/ScriptManager)
Download USCS binary bedGraphToBigWig
You will need to download the precompiled binary for converting bedGraphs to BigWig files. Navigate to UCSC executables FTP. Click the link to the OS that matches your machine and scroll down to download the bedGraphToBigWig binary.
- Linux/MacOS
Make sure you modify the permissions so that you can execute the binary. You can do this with chmod
chmod 755 /path/to/bedGraphToBigWig
For example, if your bedGraphToBigWig binary downloaded to your Downloads folder,
chmod 755 ~/Downloads/bedGraphToBigWig
Download data
You need one file of sequencing data alignments (BAM) to complete this exercise and a file with the sacCer3 yeast reference genome chromosome sizes (chrom.sizes). Read more about the BAM file format here.
BAM File
This is the set of Reb1 read alignments from the Yeast Epigenome Project (YEP). See Rossi et al (2021) for more details.
Download sample BAM fileOR

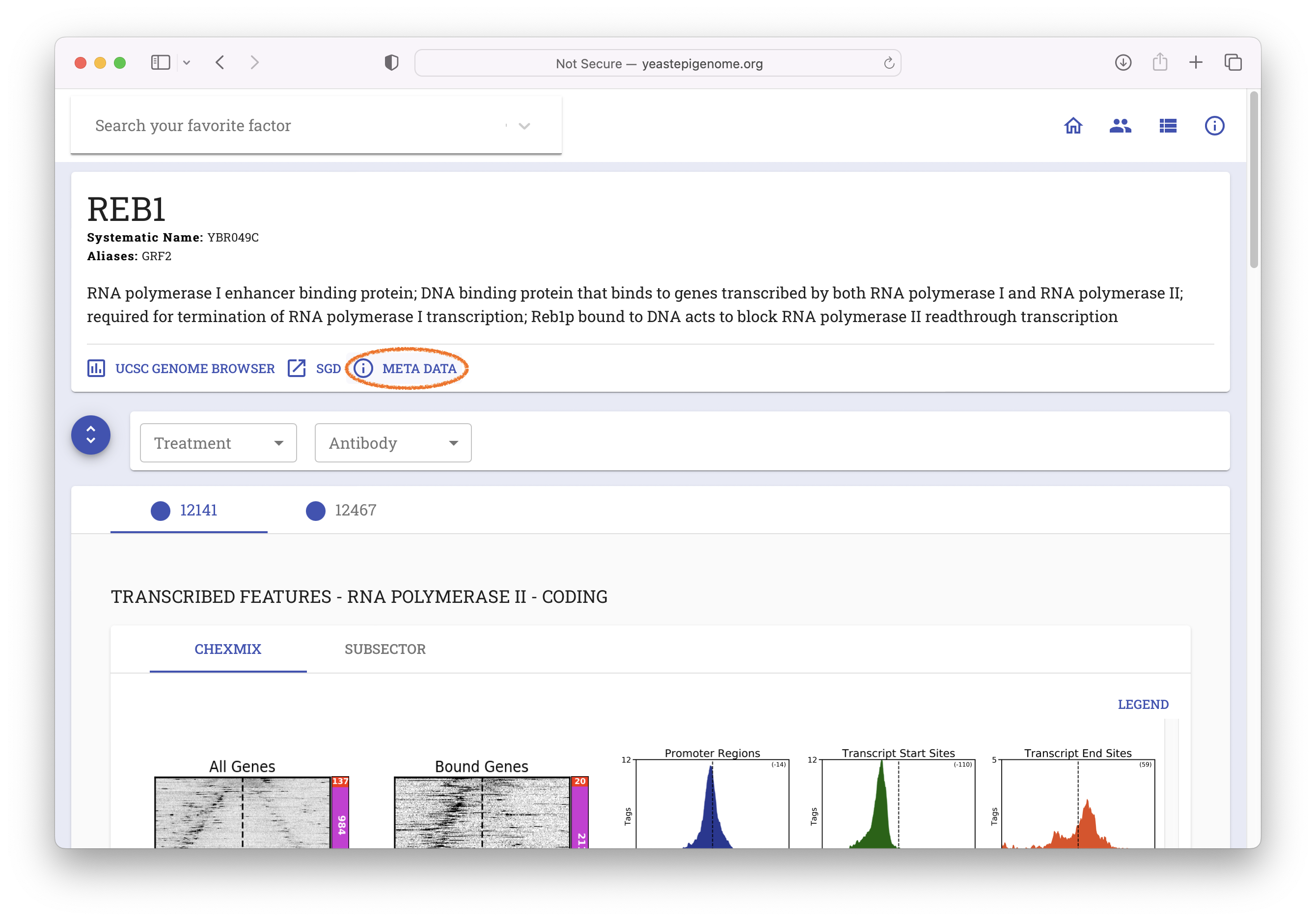
- Navigate to www.yeastepigenome.org and search for Reb1
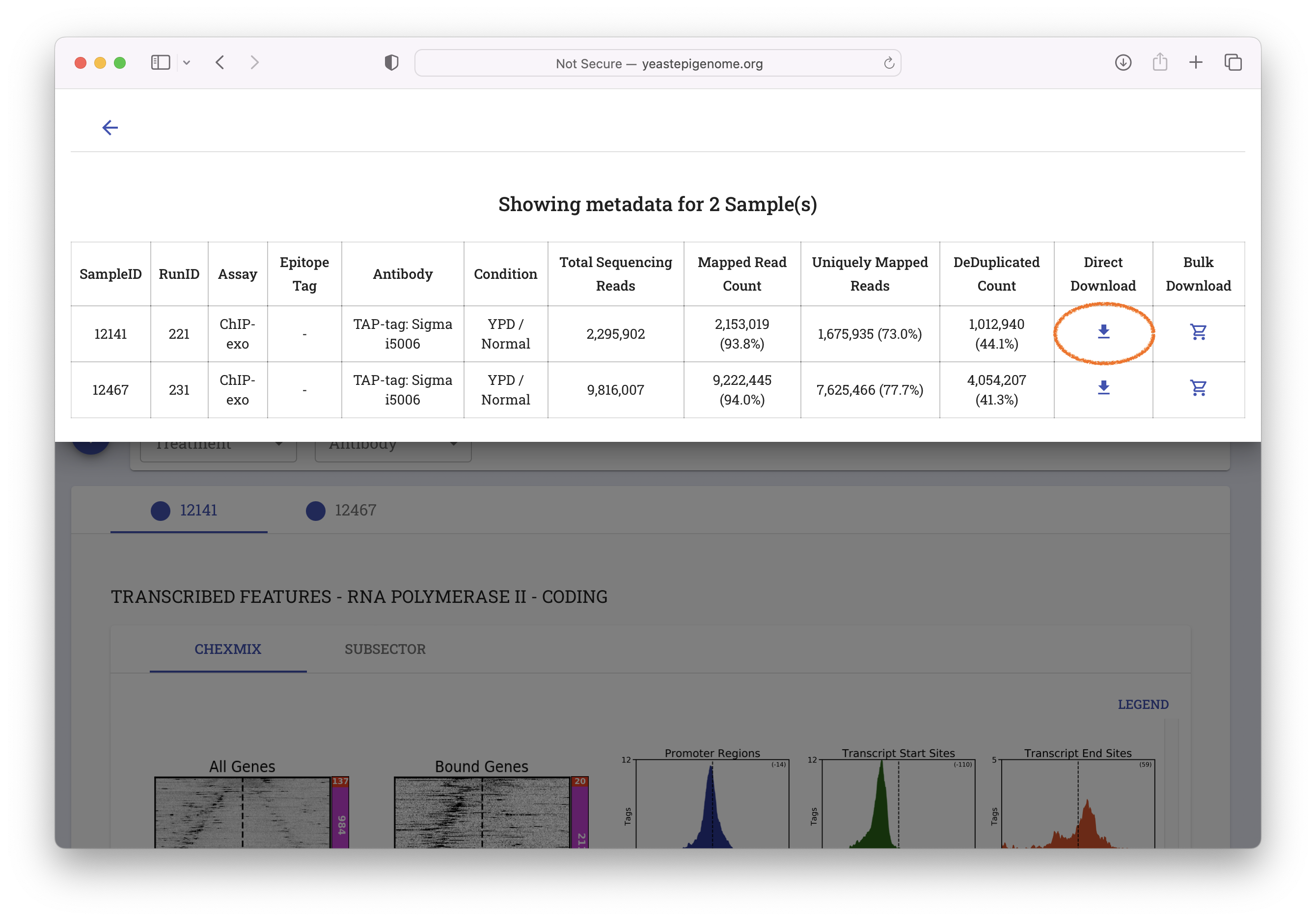
- Select "META DATA"
- Select "Direct Download"
- Unzip the resulting file ‘12141_YEP.zip’ and inspect the contents of the new
12141_YEPfolder. It should contain a file called12141_filtered.bam.
XXXX.chrom.sizes Reference File
You will also need the chromosome sizes file of the yeast reference genome (sacCer3):
Download sacCer3.chrom.sizes (TXT)The downloaded sacCer3.chrom.sizes file may be using the roman numeral naming system (chrI chrII chrIII ...). You will need to modify this to the arabic (chr1 chr2 chr3 ...) numeral naming system if you plan to use the BAM file linked above. You can do this in a simple text editor.
Also, if the file is missing the 2-micron row, you will need to add it manually. Simply append the following tab-delimited row to the bottom of the sacCer3.chrom.sizes file.
2-micron 6318
Generate the Genome Tracks
1. Open ScriptManager
- MacOS
- Linux
- Windows
Depending on your system permissions, you may need to be an administrator to open this for the first time. On Mac systems, this can be done by right-clicking the file and selecting ‘Open’ at the top.
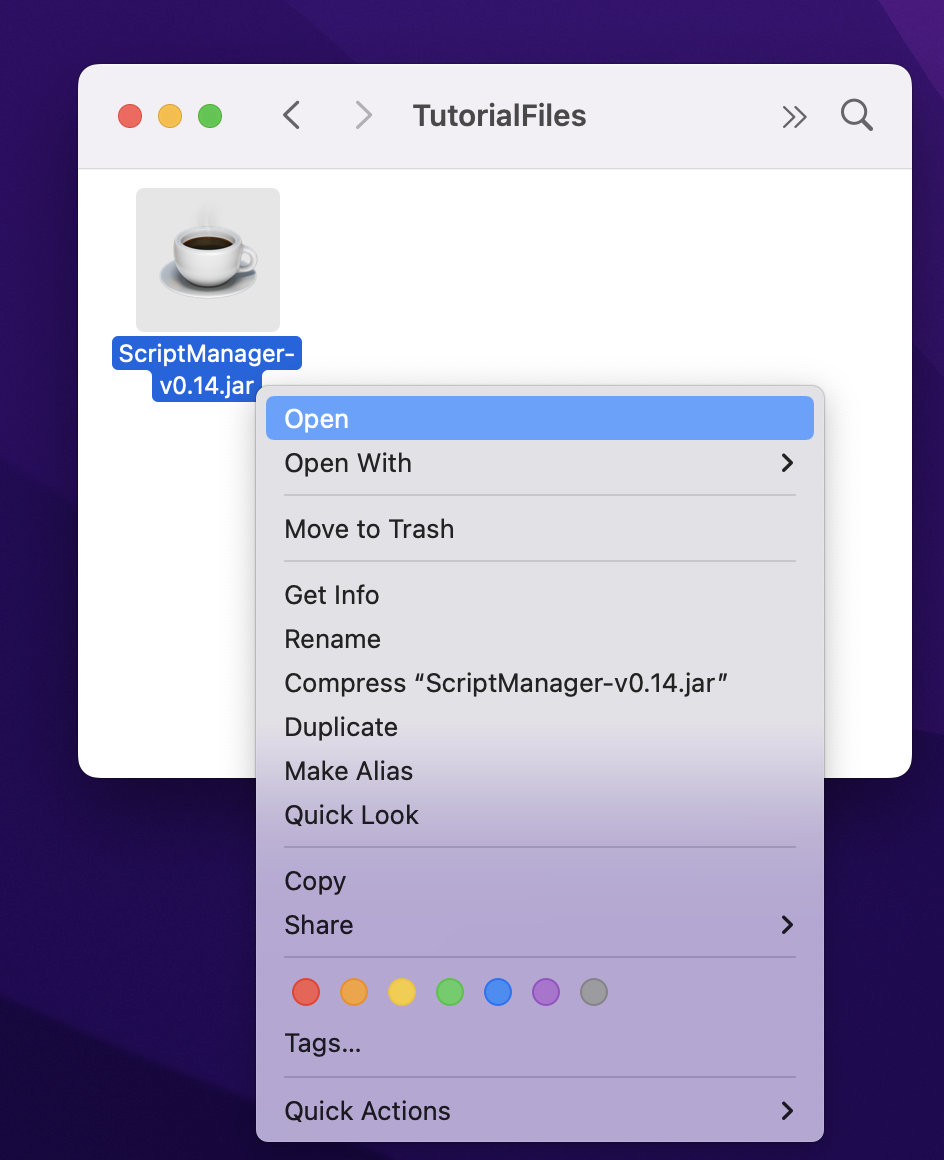
Some MacOS systems may not properly open the JAR by simply double-clicking on the JAR file so you may need to open your Terminal window and execute it from the command line by executing the jar file without any arguments or flags:
java -jar /path/to/ScriptManager.jar
If you're not sure how to type the path to ScriptManager, you can type java -jar (end with space) and then drag ScriptManager from Finder into your Terminal window and then press enter.
Double-click or right-click the ScriptManager JAR file to start the program.

Double-click or right-click the ScriptManager JAR file to start the program.

Once you see the main tool selection window, you're off to the races!
2. Create a whole-genome pileup using BAM Format Converter Tools
2.1. Navigate to BAM Format Converter ➡️ BAM to scIDX to do a genome-wide pileup of your BAM
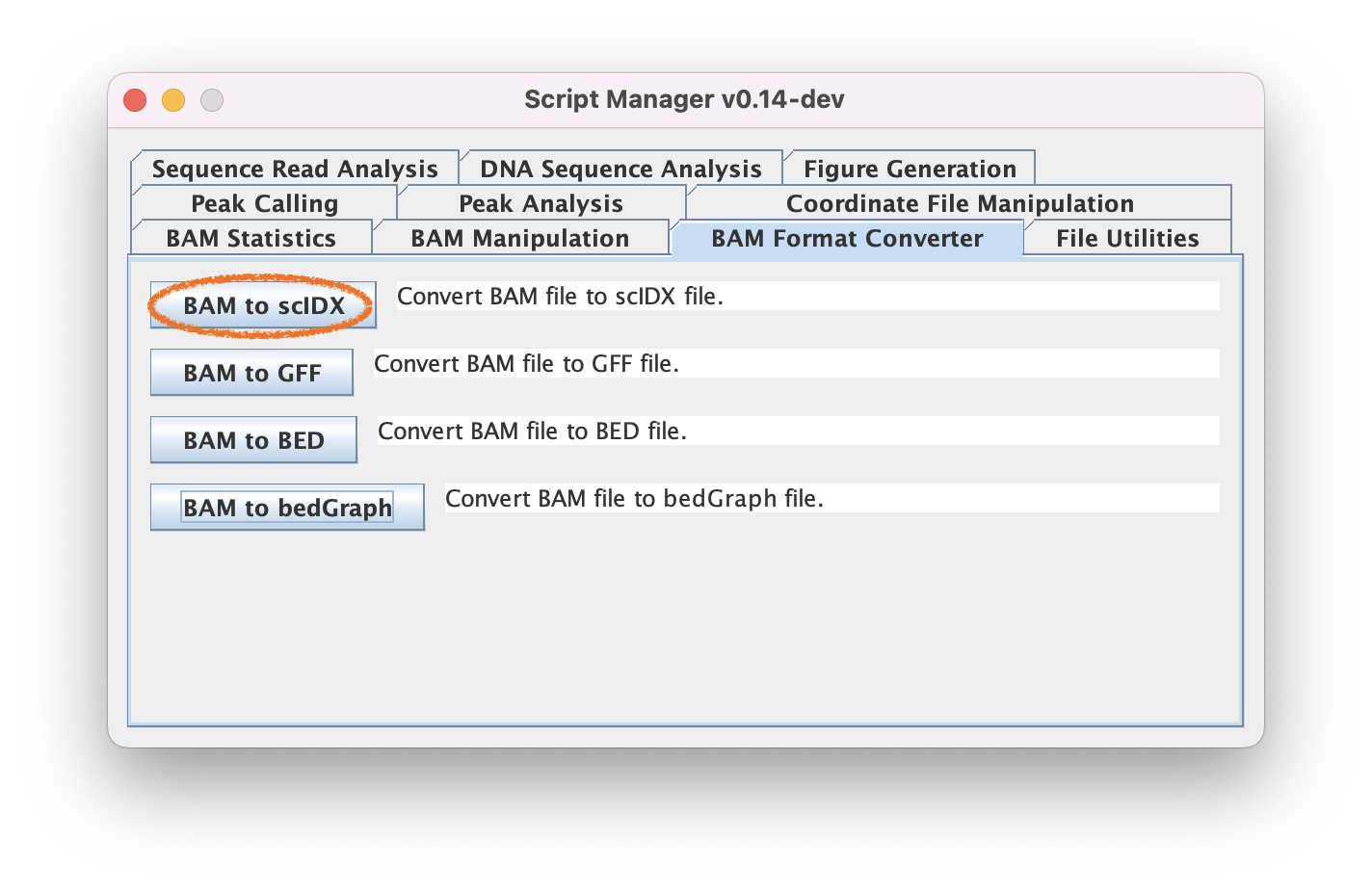
2.2. Select Encoding information
Since this is a ChIP-exo dataset, we will select Read 1 (5' end) for the encoding. You may select insert size filters if you wish.
2.3. Execute pileup
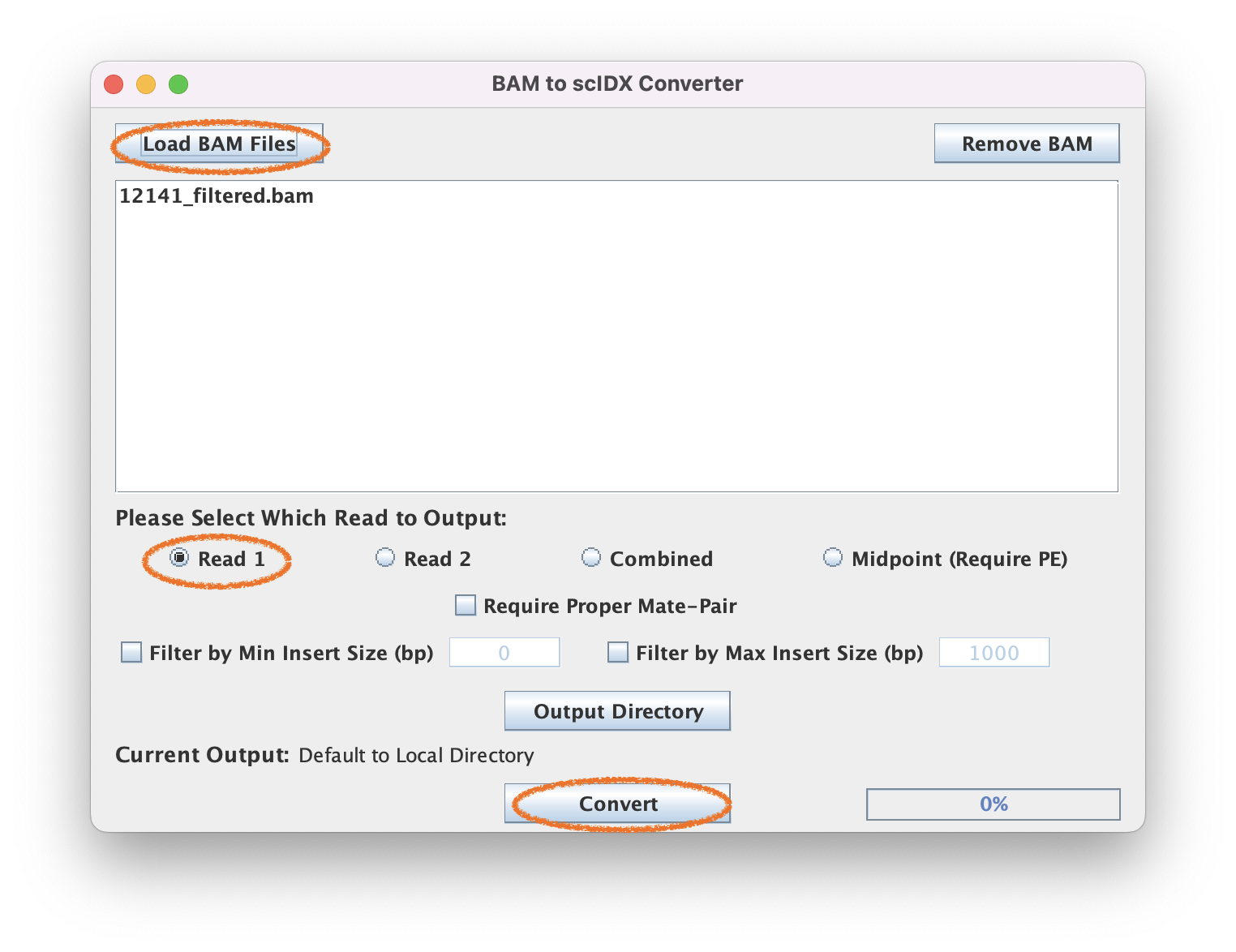

This should output a 12141_filtered_READ1.tab file.
3. (Optional) Normalize the genome track
Skip this if you would like to make a track of the raw read counts.
ScriptManager includes a "Calculate Scaling Factor" tool under the "Read Analysis" tool group that can calculate scaling factors based on several methodologies including total tag normalization and the Normalization of ChIP-seq (NCIS) protocol. Read more in the docs. Please be sure to check that your normalization method is appropriate for your data.
3.1. Navigate to Read Analysis ➡️ Calculate Scaling Factor to calculate a scaling factor
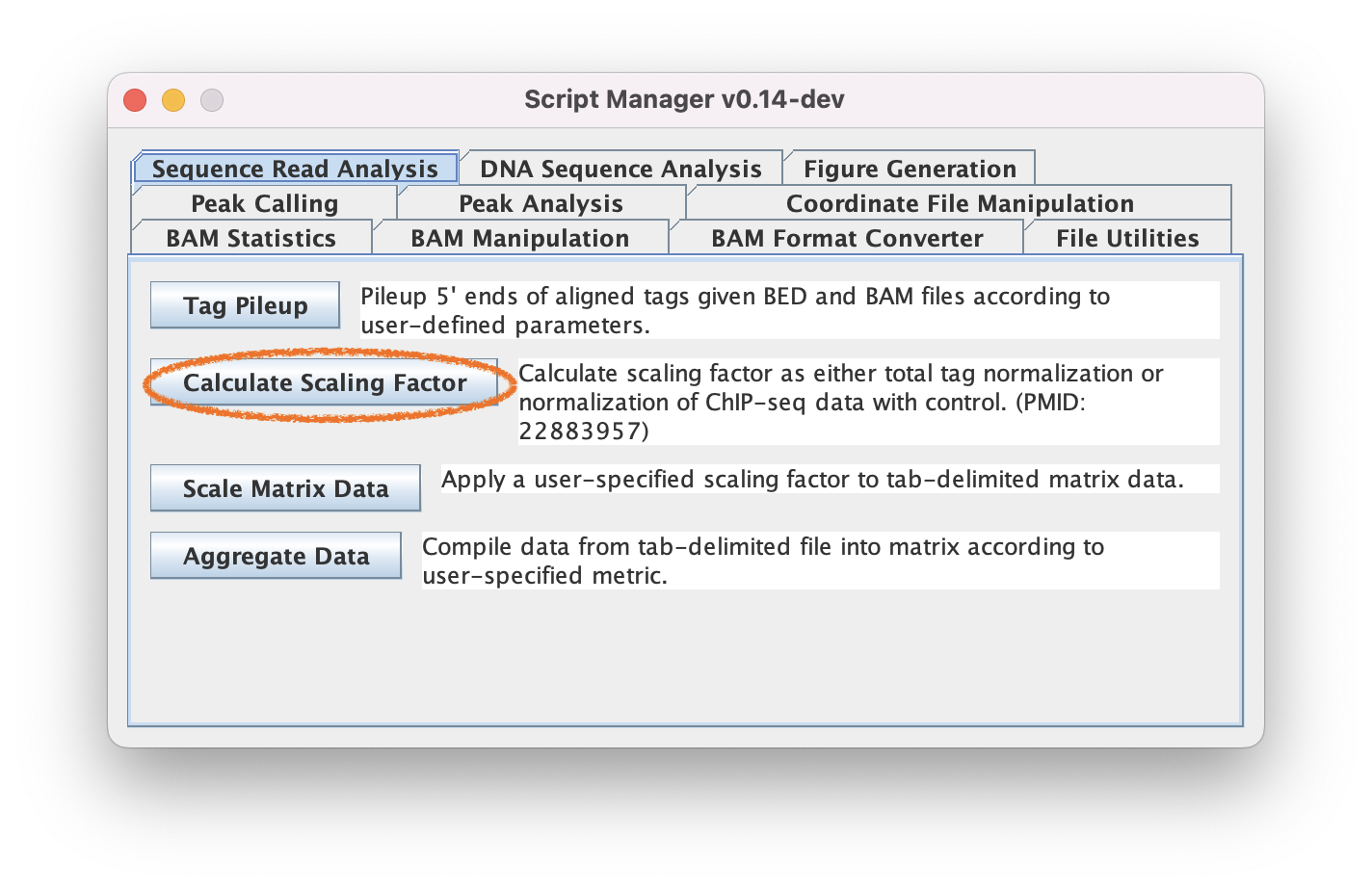
3.2. Select the BAM file you want to create the data track from
3.3. Select a normalization method and execute
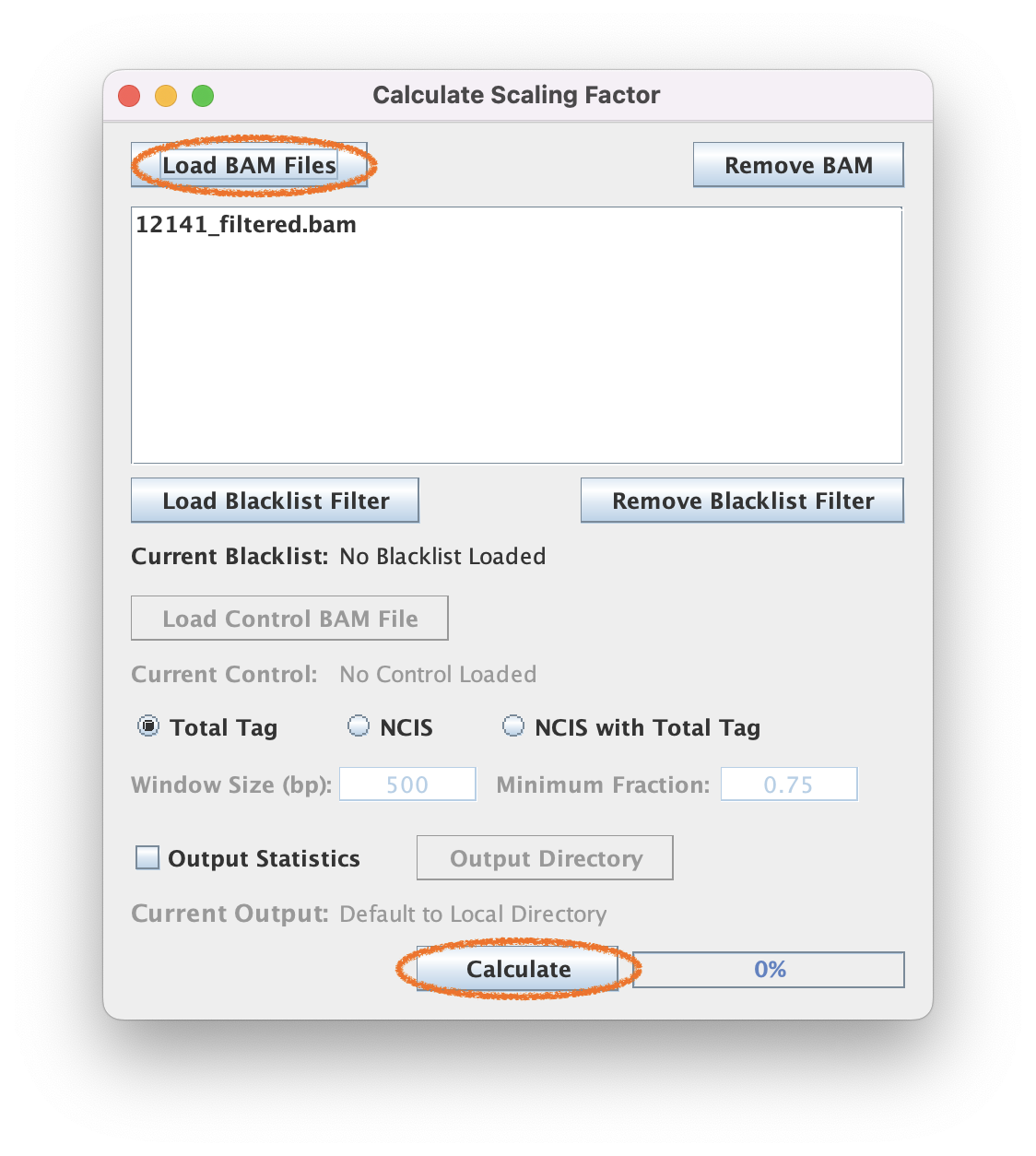
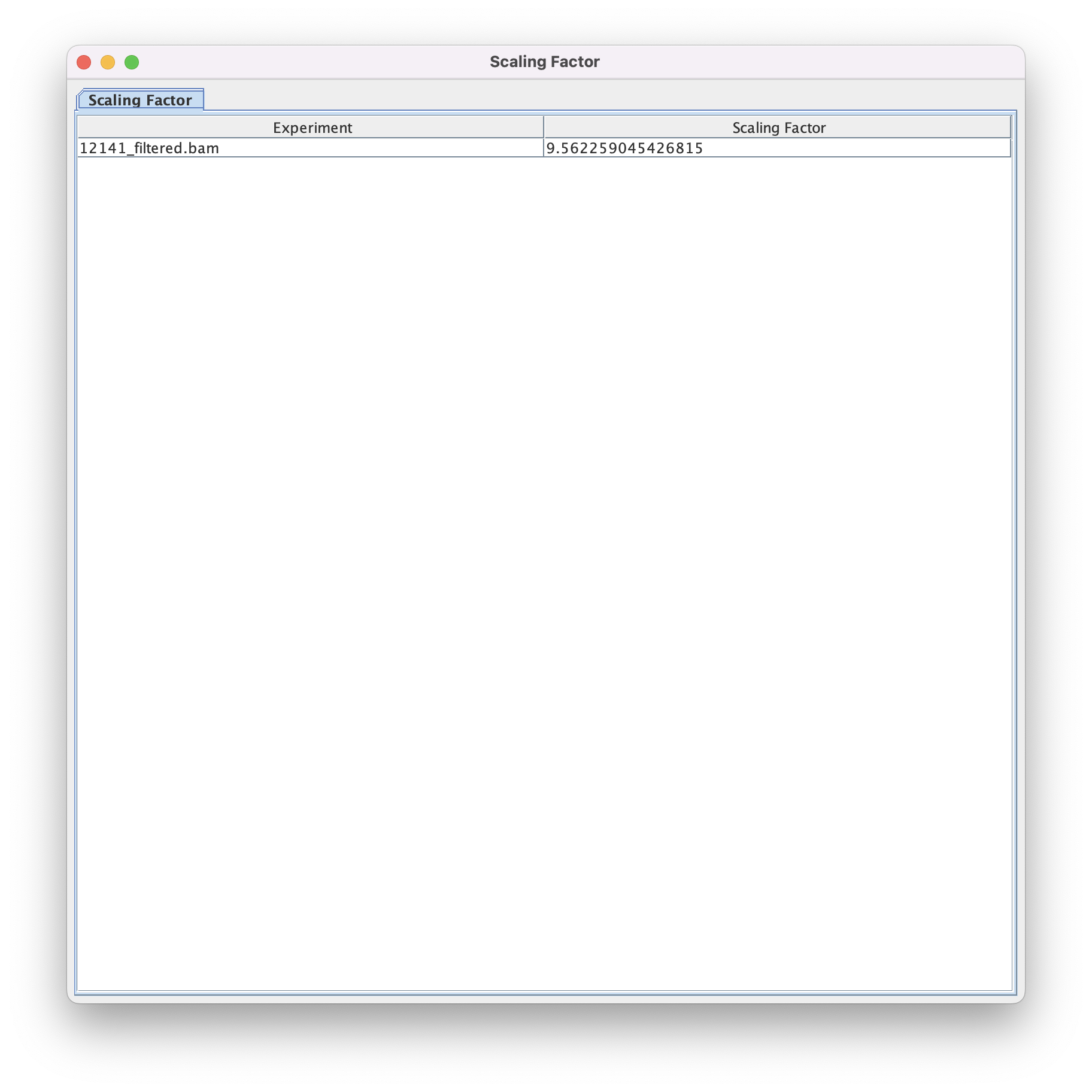

For this BAM file, the total tag method gives a scaling factor of 9.562259045426815.
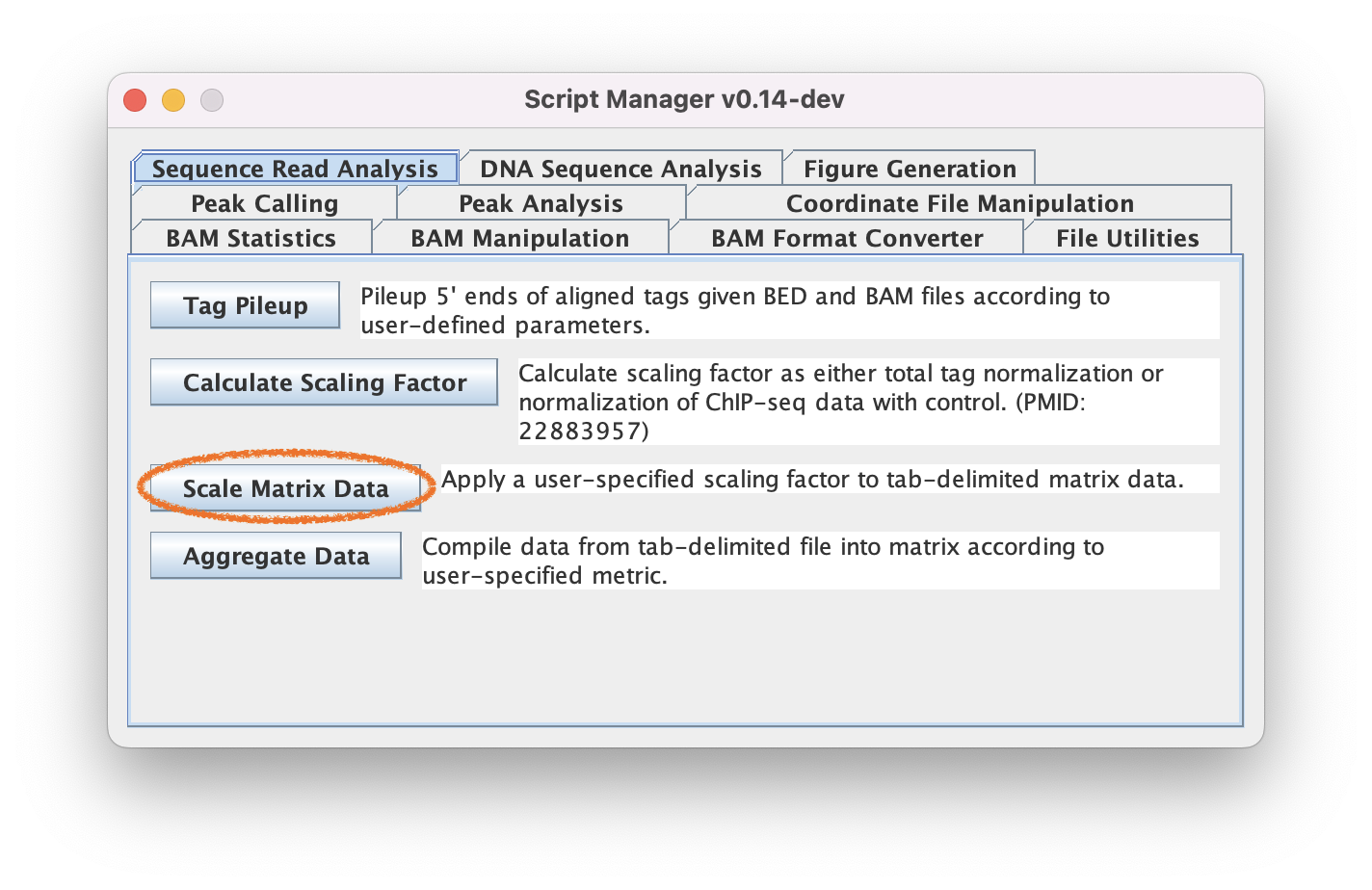
3.4. Navigate to Read Analysis ➡️ Scale Matrix to apply the normalization to the data track
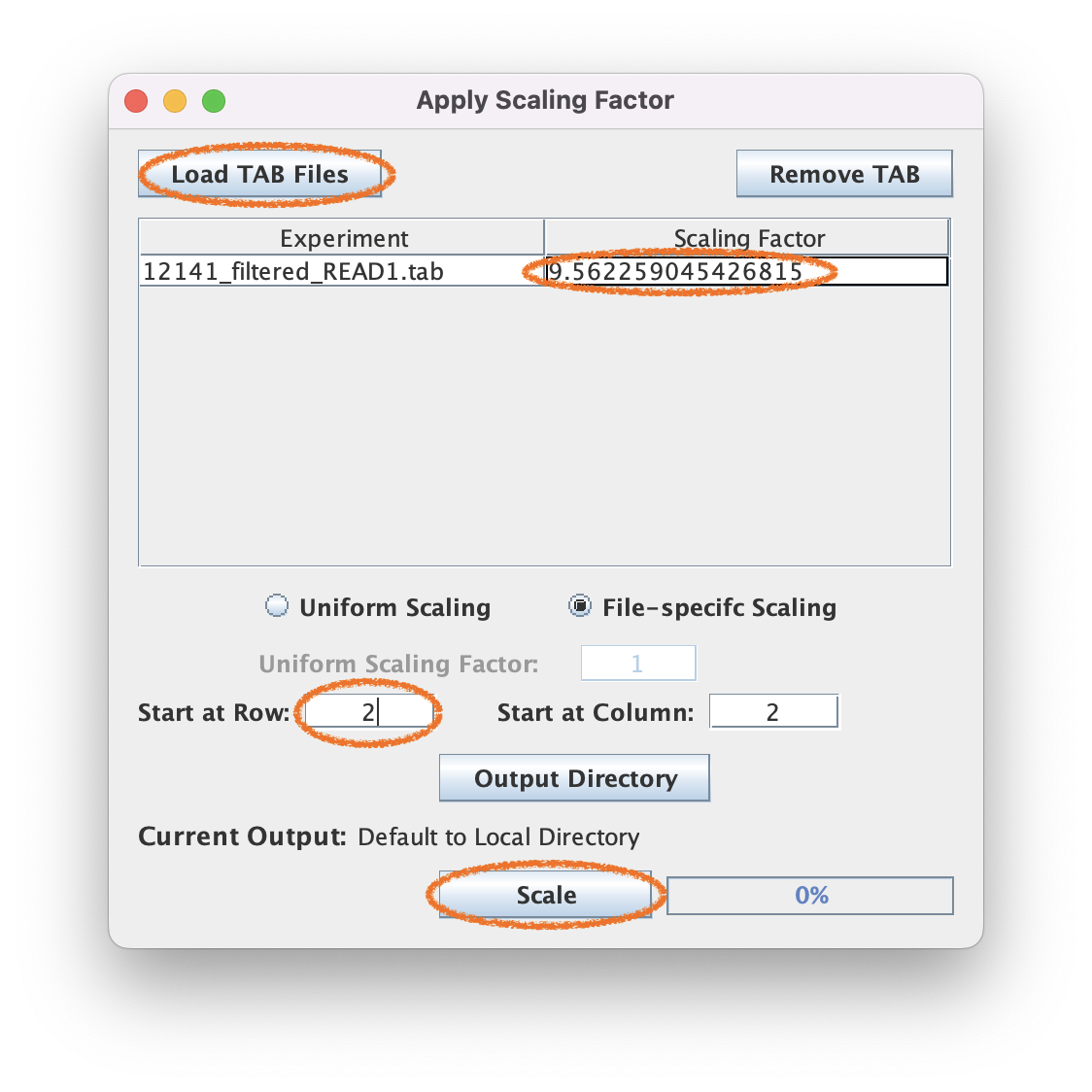
3.2. Input the normalization factor and adjust the start row to 2
3.3. Load the file and execute matrix scaling
You will be loading in the scIDX file (not the BAM).
This should output a 12141_filtered_READ1_SCALE.tab file.
4. Convert scIDX to BigWig
Since ScriptManager currently does not support converting BedGraph files to BigWigs, this step must be done in your terminal/on the command-prompt.
4.1. Turn your scIDX file into a bedGraph file
- Shell
- Excel
grep -v -f sacCer3.chrom.sizes 12141_filtered_READ1_SCALE.tab > 12141_filtered_READ1_SCALE.filtered.tab
sed '1d;2d' 12141_filtered_READ1_SCALE.filtered.tab | awk '{OFS="\t"}{FS="\t"}{print $1, $2, $2+1, $3}' |sort -k1,1 -k2,2n > 12141_filtered_READ1_SCALE_forward.bedgraph
sed '1d;2d' 12141_filtered_READ1_SCALE.filtered.tab | awk '{OFS="\t"}{FS="\t"}{print $1, $2, $2+1, $4}' |sort -k1,1 -k2,2n > 12141_filtered_READ1_SCALE_reverse.bedgraph
Open your file in excel and strip the file of the first two rows:
#2023-03-10 12:52:40.439;12141_filtered.bam;READ1
chrom index forward reverse value
You may also have read mapping to the very edges of the chromosome. These entries will also have to be removed.
Then rearrange the columns into BedGraph format by copying them in the following order (3rd column is the value of col2 plus one):
12141_filtered_READ1_SCALE_forward.bedGraph: col1, col2, (col2 +1), col312141_filtered_READ1_SCALE_reverse.bedGraph: col1, col2, (col2 +1), col4
...and then sort each of them by the first and then the second columns. This format is tab-delimited (not comma-delimited!) so save as a tab-delimited text file. Do not save this as an Excel file format (.xlsx, .xls, etc). This should be a .tsv or .tab format.
4.2. Turn your bedGraph file into a BigWig file
- Shell
./bedGraphToBigWig 12141_filtered_READ1_SCALE_forward.bedGraph sacCer3.chrom.sizes my-track-basename_forward.bw
./bedGraphToBigWig 12141_filtered_READ1_SCALE_reverse.bedGraph sacCer3.chrom.sizes my-track-basename_reverse.bw
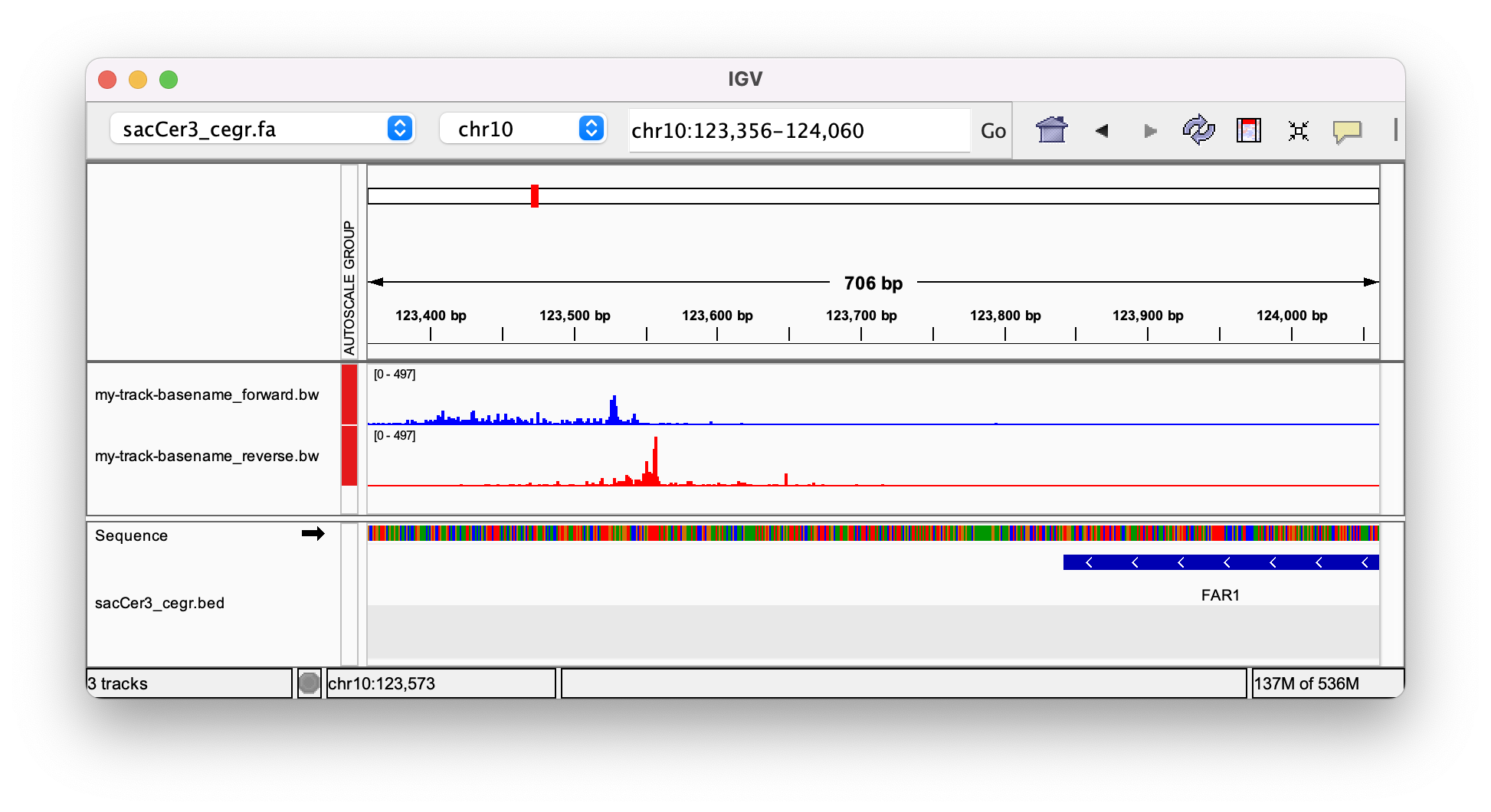
4.3. Load the BigWig into your favorite genome browser
Tah dah! You've made the genome tracks for a ChIP-exo dataset!
Command-Line shell script
The following shell commands mirror the manipulations described above to create BigWig genome tracks. This can serve as a template for you to write out your own workflows as bash scripts that execute command-line style ScriptManager.
SCRIPTMANAGER=/path/to/ScriptManager.jar
BGTOBW=/path/to/bedGraphToBigWig
CHRMSZ=/path/to/sacCer3.chrom.sizes
BAMFILE=/path/to/12141_filtered.bam
OUTPUT=/path/to/my-track-basename
FACTOR=9.562259045426815 # calculate this separately
# Index BAM if not already indexed
[ -f $BAMFILE.bai ] || samtools index $BAMFILE
# Pileup BAM along whole genome
java -jar $SCRIPTMANAGER bam-format-converter bam-to-scidx $BAMFILE -o $OUTPUT.raw.tab
# Only include pileups from chromosomes in the sacCer3.chrom.sizes reference
grep -v -f $CHRMSZ $OUTPUT.raw.tab > $OUTPUT.filtered.tab
# Scale SCIDX output (optional)
java -jar $SCRIPTMANAGER read-analysis scale-matrix $OUTPUT.filtered.tab -s $FACTOR -r 2 -o $OUTPUT.scaled.tab
# Convert to forward/reverse Bedgraph tracks
sed '1d;2d' $OUTPUT.scaled.tab | awk '{OFS="\t"}{FS="\t"}{print $1, $2, $2+1, $3}' |sort -k1,1 -k2,2n > $OUTPUT.forward.bedgraph
sed '1d;2d' $OUTPUT.scaled.tab | awk '{OFS="\t"}{FS="\t"}{print $1, $2, $2+1, $4}' |sort -k1,1 -k2,2n > $OUTPUT.reverse.bedgraph
# Compress to BigWig format
$BGTOBW $OUTPUT.forward.bedgraph $CHRMSZ $OUTPUT\_forward.bw
$BGTOBW $OUTPUT.reverse.bedgraph $CHRMSZ $OUTPUT\_reverse.bw
# Clean-up
rm $OUTPUT.raw.tab $OUTPUT.filtered.tab $OUTPUT.scaled.tab $OUTPUT.forward.bedgraph $OUTPUT.reverse.bedgraph
# Output files:
# - /path/to/my-track-basename_forward.bw
# - /path/to/my-track-basename_reverse.bw