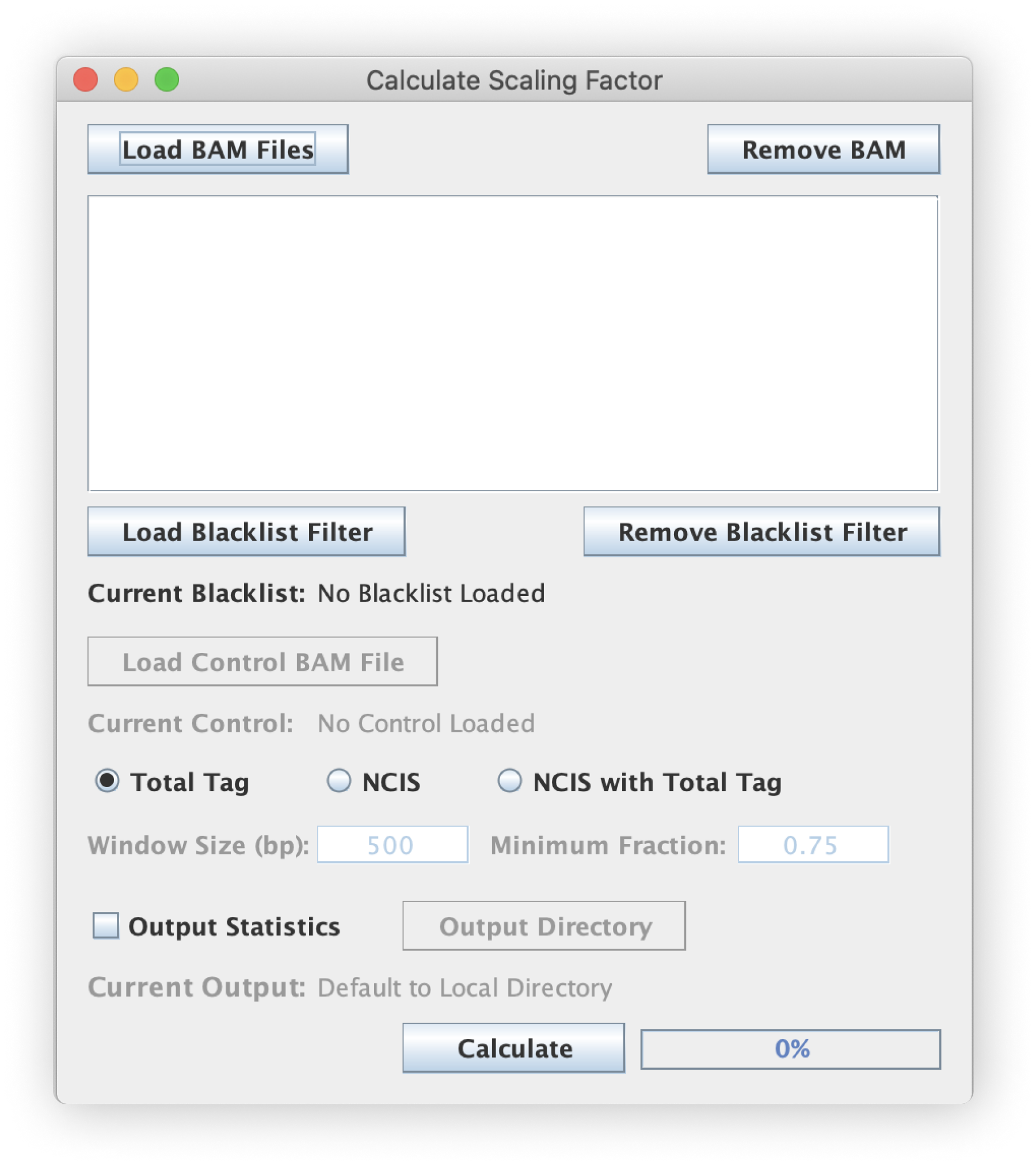
Scaling Factor

Calculate a normalization factor using the total tag method or normalization of ChIP-seq data with control (NCIS) method by Liang & Keles (BMC Bioinf 2012).
The resulting scaling factor can be applied to read count matrices generated by Tag Pileup using Scale Matrix to compare experiment samples.
File input (BAM)
The scaling factors are calculated directly from a BAM file and are calculated on a per-BAM basis. Make sure your input is properly formatted and uses the appropriate .bam extension. The scaling factor determined for one file should not be used to normalize data from another BAM file.
Filter
- The Blacklist option allows the user to provide a file containing blacklisted entires to exclude certain regions of data from the calculations. This commonly includes known repetitive/problematic mapping regions of the genome.
- There are also options specific for when the user selects an NCIS-style calculation method:
- The Control BAM option is for specifying the control BAM file to model the background-signal
- The Window Size(bp) adjusts the size of the tiling window used
- The Minimum Fraction sets the minimum fraction for the NCIS method thresholding
NCIS control samples should have sufficient sequencing depth to create a reliable sampling of the genome-wide distribution of background read coverage. It's a common mistake to use under-sequenced IgG controls in ChIP-based analyses.
Scaling Methods
- Total Tag normalization
- Perhaps the most intuitive normalization approach, ScriptManager's Total tag normalization method simply tallies up the reads (only Read 1 if paired-end) in a BAM file and divides it by the genome size for a more intuitive "per bp" coverage metric. This has the added advantage of avoiding underflow errors when the factor is applied to a read count matrix (e.g. CDT output file from TagPileup).
Note that the genome size is determined by the BAM header so be careful about comparing BAM files that were aligned to the exact same reference genome.
- NCIS normalization
- Pros: Attempts to account for antibody specificity
- Cons: Requires IgG control (but this is best practice anyway to avoid false positive peak calls)
- Read more
NCIS is not an appropriate normalization method for ChIP data with histone targets (violates assumptions of sparse binding). Consider NFR normalization approaches for histone target data (but understand assumptions before proceeding).
- Both total tag and NCIS methodologies
Command Line Interface
Usage:
java -jar ScriptManager.jar read-analysis scaling-factor [-t | -n | -b] [-hV]
[-c=<controlBAM>] [-f=<blacklistFilter>] [-m=<minFrac>]
[-o=<outputBasename>] [-w=<window>] <bamFile>
Filter Options
| Option | Description |
|---|---|
-f, --blacklist=<blacklistFilter> | specify blacklist file to filter by |
-c, --control=<controlBAM> | control BAM file (to use with -n or -b flags) |
-w, --window-size=<window> | window size for NCIS-related scaling types (default=500) |
-m, --min-fraction=<minFrac> | minimum fraction for NCIS-related scaling types (default=0.75) |
Positional Input
This tool takes a single BAM file for input. As with other tools, this tool requires the BAM file be indexed.
Output Options
| Option | Description |
|---|---|
-o, --output=<outputBasename> | specify output file for composite values |
Scale Options
| Option | Description |
|---|---|
-t, --total-tag | total tag scaling (default) |
-n, --ncis | ncis normalization with window size in bp and unitless minimum fraction (default-size=500, default-fraction=0.75) |
-b, --both | ncis with total tag (default-size=500, default-fraction=0.75) |