FRiX Score Calculator
Quantify library enrichment around a set of Reference points (Fraction of Reads in Peak/Motif, aka FRiP/FRiM).
Evaluating enrichment of a genomic library around a given set of annotations is a useful way to check how much signal is in a genomic library like a ChIP-seq dataset.
File inputs (BAM & BED)
This script processes BAM-type files so make sure your input is properly formatted and uses the appropriate .bam extension.
This script also processes BED-type files so make sure your input is properly formatted and uses the appropriate .bed or .bed.gz extension.
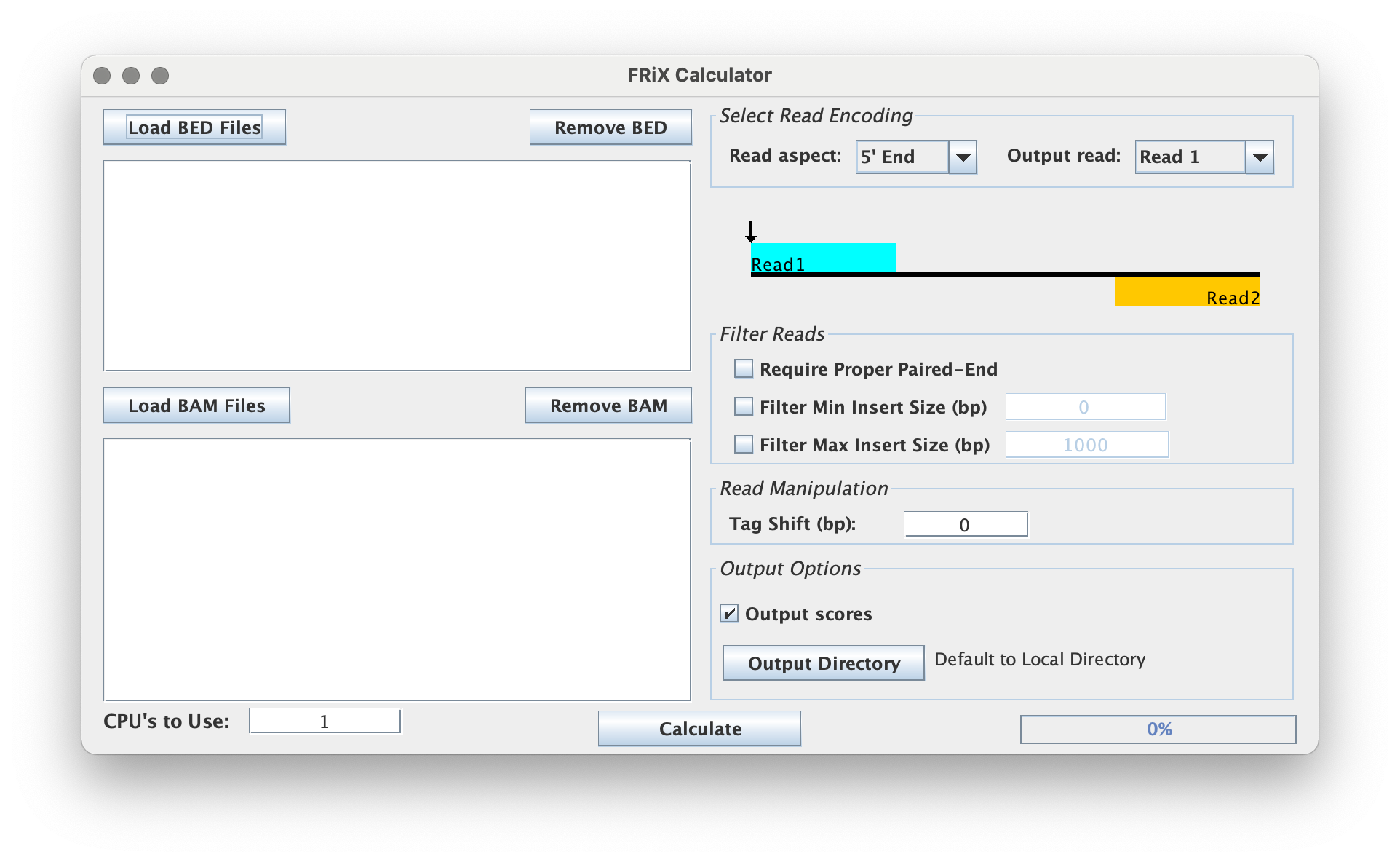
Read Aspect & Type
This tool has multiple read aspects to choose from.
- 5' End: analyze the 5' end of the given read
- 3' End: analyze the 3' end of the given read
- Midpoint: analyze the midpoint between two reads
Note: The Midpoint option requires proper paired-end reading.
For the 5' End and 3' End options, the tool also provides the selection to analyze Read 1, Read 2, or All Reads.
Filter Options
- Require Proper Paired-End refers to the proper pairing of reads Read 1 and Reads 2.
- Filter Min Insert Size (bp) refers to the minimum insert size to filter
- Filter Max Insert Size (bp) refers to the maximum insert size to filter
Read Manipulation
The user can shift the aligned tags by indicating the number of base pairs to be shifted by in the 'Tag Shift' box.
Output Statistics
Below is an example of the text output file which shows the set of metrics calculated.
BAM filename: /path/to/MyBAMFile.bam
RefPT filename: /path/to/MyRefPT_100bp.bed
Number of Sites: 69733
Total aligned read count: 9435384.0
Total genome size: 3.137161264E9
Summed tags at all sites: 19851.0
FRiX score: 0.0021038889355218613
FRiX density: 6.706346146960016E-13
Command Line Interface
Usage:
java -jar ScriptManager.jar peak-analysis frix-score [-5 | -3 | -m] [-1 | -2 | -a | -m] [-dhptVz] [--cpu=<cpu>] [-n=<MIN_INSERT>][-s=<shift>] [-x=<MAX_INSERT>] [-o=<output>] <bedFile> <bamFile>
Positional Inputs
| Option | Description |
|---|---|
bedFile | The BED file with reference coordinates to pileup on. |
bamFile | The BAM file from which we remove duplicates. Make sure its indexed! |
Read Options
| Option | Description |
|---|---|
-1, --read1 | pileup of read 1 (default) |
-2, --read2 | pileup of read 2 |
-a, --all-reads | pileup all reads |
-m, --midpoint | pile midpoint (require PE) |
Calculation Options
| Option | Description |
|---|---|
-s, --shift=<shift> | set a shift in bp (default=0bp) |
-t, --standard | set tags to be equal (default=false) |
--cpu=<cpu> | set number of CPUs to use (default=1) |
Filter Options
| Option | Description |
|---|---|
-p, --require-pe | require proper paired ends (default=false), automatically turned on with any of flags -mnx |
-n, --min-insert=<MIN_INSERT> | filter by minimum insert size in bp, require PE (default=no minimum) |
-x, --max-insert=<MAX_INSERT> | filter by maximum insert size in bp, require PE (default=no maximum) |
Output Options
| Option | Description |
|---|---|
-o, --output=<output> | specify output file |