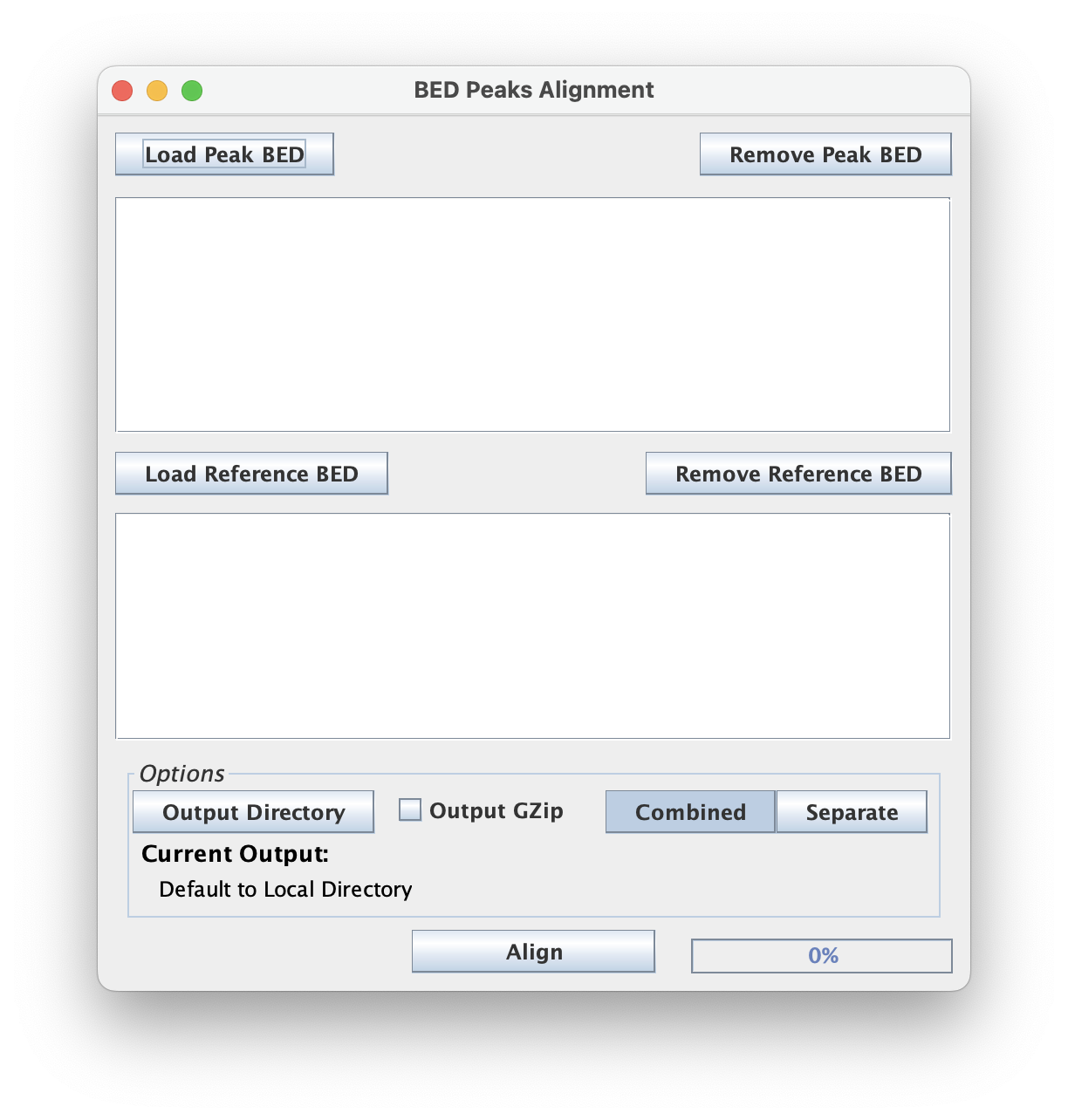
Align BED to Reference

Align BED peaks to Reference BED file creating CDT files for heatmap generation.
You can think of this like a "Tag Pileup" for annotations (BED) signal in place of the BAM signal that Tag Pileup uses.
File Inputs
This script expects both a Peak BED file input and a Reference BED file input.
Peak BED
The "Peak BED" files are a set of genomic annotations that serve as the signal to positionally tally across each record in the "Reference BED" file. These are typically peaks, motif instances, genome annotation features, or any set of genomic regions that can be represented in the BED format.
Reference BED
The "Reference BED" files are the set of genomic ranges/intervals to align the "Peaks" against.
Strand Output Options
Combined Strand option combines peaks from the antisense and snese strand into a single output, creating a unified view of the aligned peaks.
Separated Strand option separates the peaks into antinsense and sense strands, providing a detailed analysis of strand-specifc peak distibutions and patterns.
Command Line Interface
Usage:
java -jar ScriptManager.jar peak-analysis peak-align-ref [-hV] [--separate
| --combined] [-o=<output>]
<peakBED> <refBED>
Positional Input
| Input | Description |
|---|---|
<peakBED> | The BED peak file |
<refBED> | The BED reference file |
Strand Options
| Option | Description |
|---|---|
--separate | select output strands as separate (default) |
--combined | select output strands as combined |
Output Options
| Option | Description |
|---|---|
-o, --output=<output> | specify output file (default=<peakBED>_<refBED>_Output.cdt) |
-z, --gzip | gzip output (default=false) |