DNA Shape from BED File

Calculate intrinsic DNA shape parameters given BED file and Genome FASTA file. Based on Roh's lab DNAshape server data.
Based on the findings from the Rohs lab (Zhou et al, 2013; Li et al, 2017), a sliding window approach using a 5bp wide window is a strong predictor of local DNA shape. Using this approach, we can predict 13 kinds of DNA shapes. The electrostatic potential of the strand is also calculable using a similar approach (Chiu et al,2017).
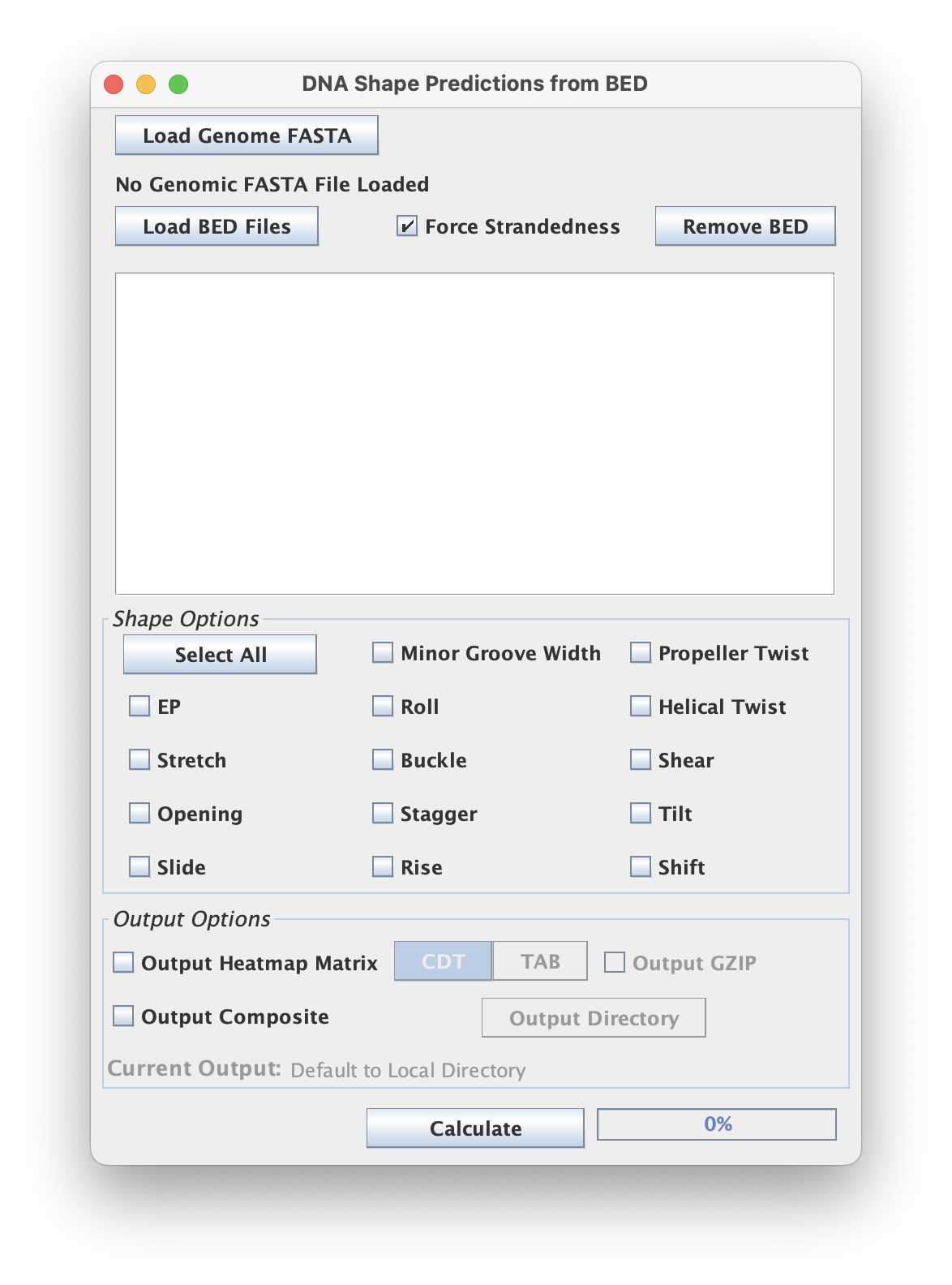
This script takes in a series of nucleotide sequences from a BED file and determines the average shape score(s) across the bp positions.
What do these shape options mean?
Below is a video introducing some of the shape measurements that we are trying to capture with these calculations.
File inputs (BED & FASTA)
The sequence for each input BED-specified coordinates in the FASTA file has a shape score series pattern. Because the shape score is a series corresponding to the bp position, the BED records in the input should be positionally linked to some feature and of the same length.
When using the GUI, make sure your input is properly formatted and uses the appropriate BED (.bed or .bed.gz) and FASTA (.fa / .fa.gz / .fasta / ...) extensions.
File Options
The 'Force Strandedness' options ensures that the DNA strand orientation during the analysis is considered.
Output file (CDT/TAB)
The average composites of the CDT output will be displayed in the GUI output window:
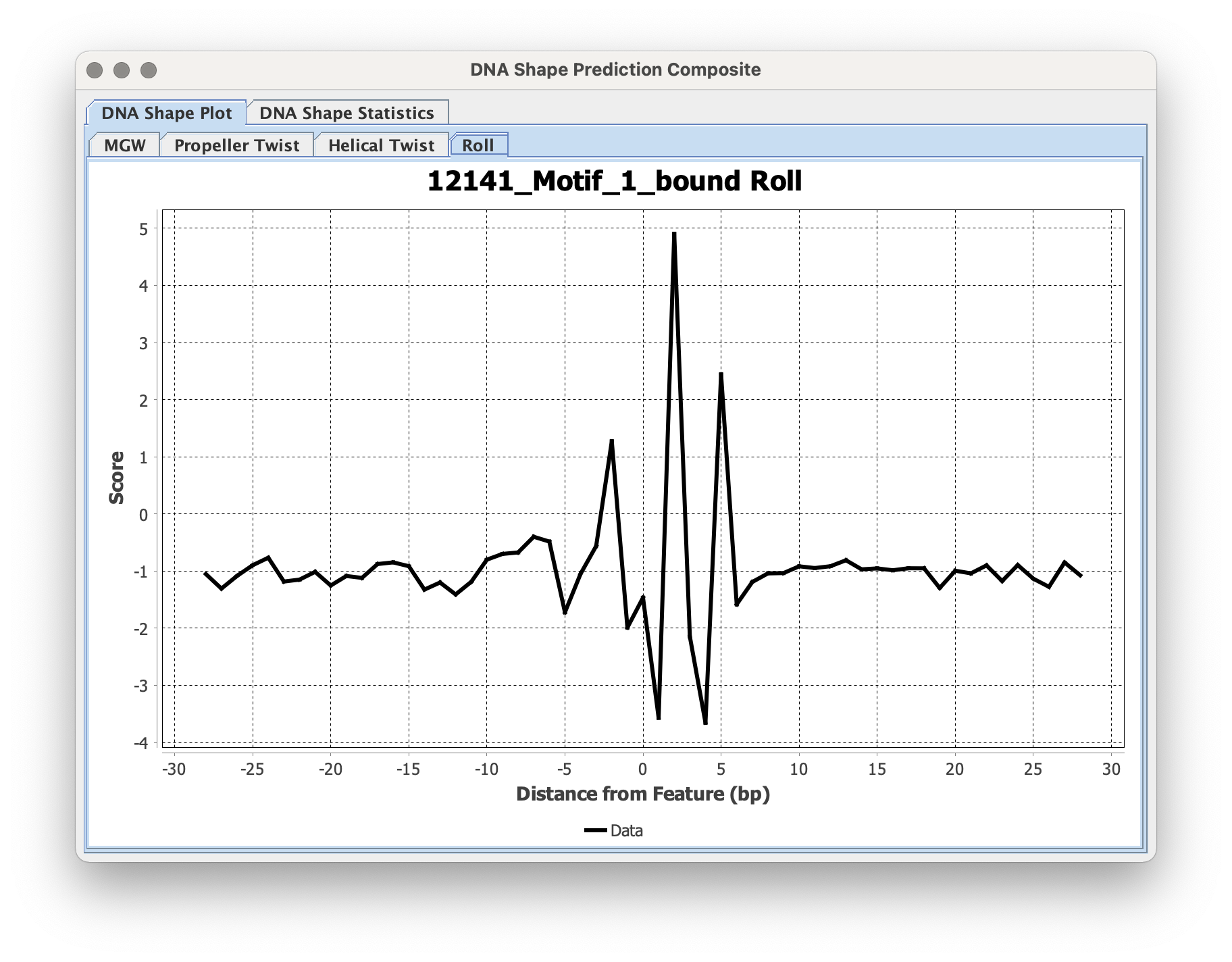

There should be a CDT file/Composite file output for each shape aspect selected based on the input filename and with a suffix distinguishing each selected shape style (_HelT.cdt, _MGW.cdt, _PropT.cdt, and _Roll.cdt).
For example, in the command-line execution, an -o myoutput argument can be provided and the resulting files should be called myoutput_MGW.cdt, myoutput_PTwist.cdt, myoutput_HTwist.cdt, or myoutput_Roll.cdt according to the shapes selected (or with .out if composite is selected).
The output matrix files use the same format as the output from Tag Pileup (can visualize with Figure Generation's heatmap and composite tools).
Command Line Interface
Usage:
java -jar ScriptManager.jar sequence-analysis dna-shape-bed [-afghlprV]
[--avg-composite] [-o=<outputBasename>] <genomeFile> <bedFile>
Based on Roh's lab DNAshape server data. Notes: Sequences with Ns are thrown out.
Positional Input
Expects a FASTA formatted file with many sequences to stack up with each other (like fasta-extract tool output).
| Option | Description |
|---|---|
<genomeFile> | reference genome FASTA file |
<bedFile> | the BED file of sequences to extract |
Output Options
| Option | Description |
|---|---|
-o, --output=<outputBasename> | Specify output basename (files for each shape indicated will share this base) |
-z, --gzip | gzip output (default=false) |
--avg-composite | Save average composite |
For each shape option to calculate indicated by the command, a CDT file will be generated with an extension indicating the shape type calculated.
If the groove information is indicated in the command to be used for the output, a file called <outputBasename>_MGW.cdt will be generated.
Similarly for propeller, helical, and roll, the output matrix CDT files will be named with the suffixes _PTwist.cdt, _HTwist.cdt, and _Roll.cdt, respectively.
Strand Options
| Option | Description |
|---|---|
-f, --force | force-strandedness (default) |
Shape Options
| Option | Description | Units | Image |
|---|---|---|---|
-g, --groove | Output minor groove width | Angstroms | 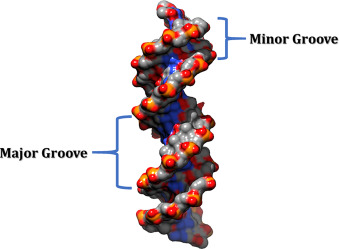 |
-r, --roll | Output roll | Degrees |  |
-p, --propeller | Output propeller twist | Degrees | 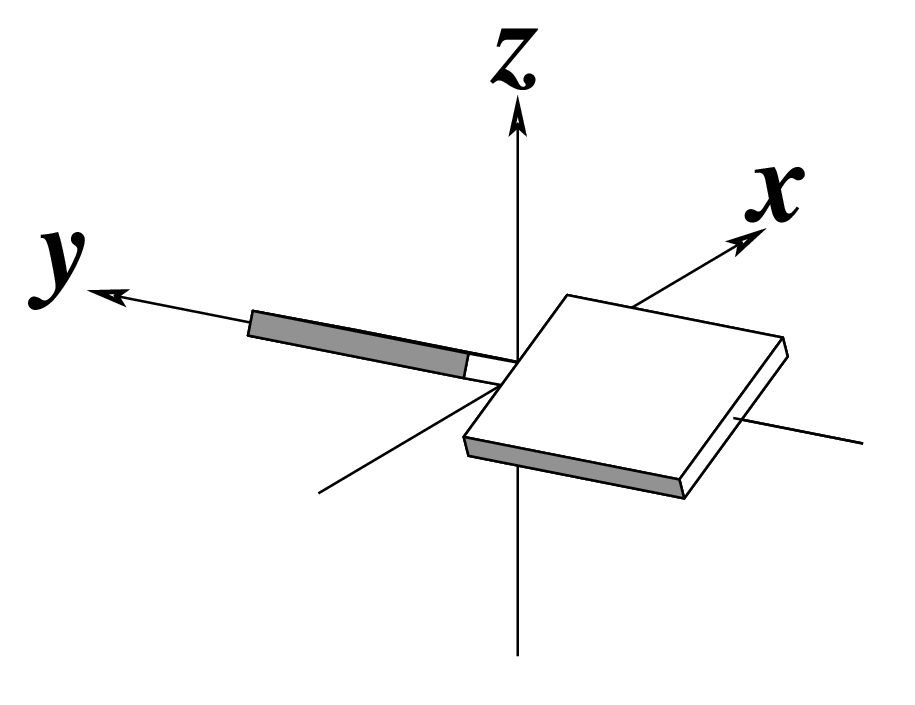 |
-l, --helical | Output helical twist | Degrees | 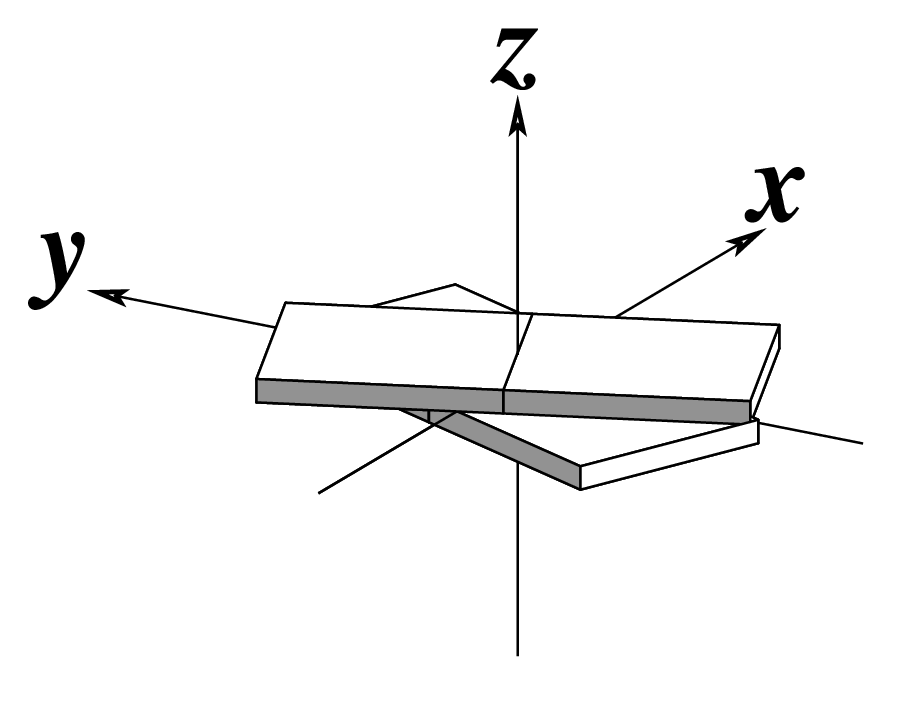 |
--electrostatic-potential | Output electrostatic potential | kT/e | 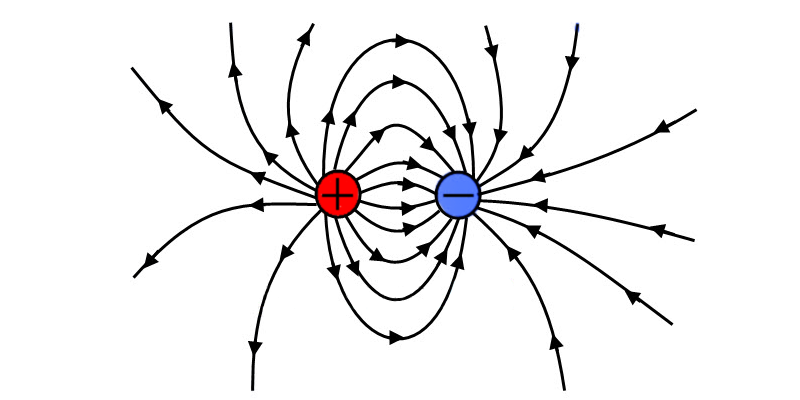 |
--stretch | Output stretch | Angstroms | 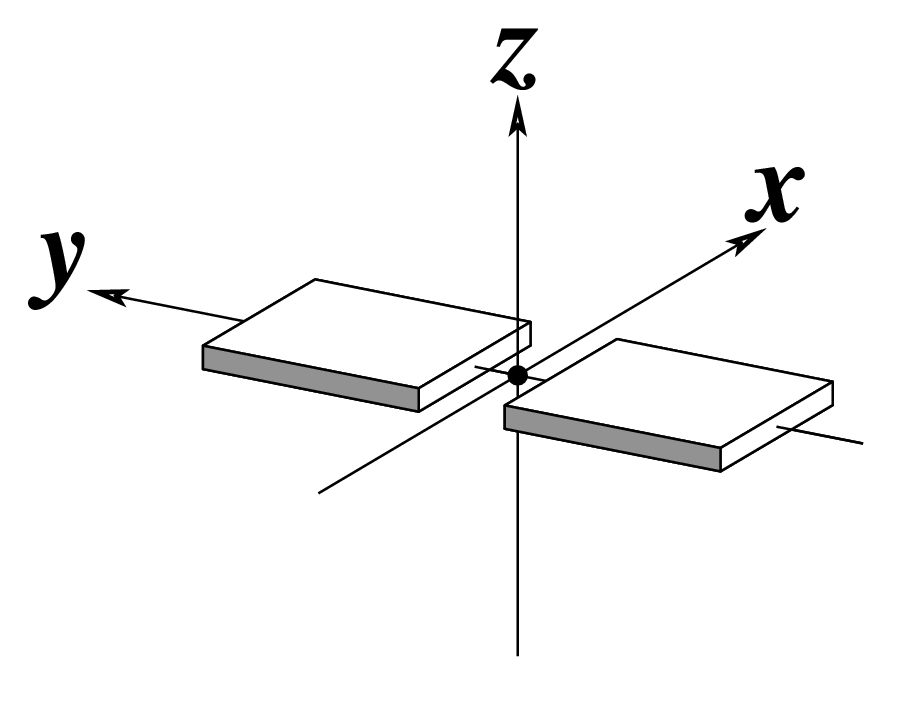 |
--buckle | Output buckle | Degrees | 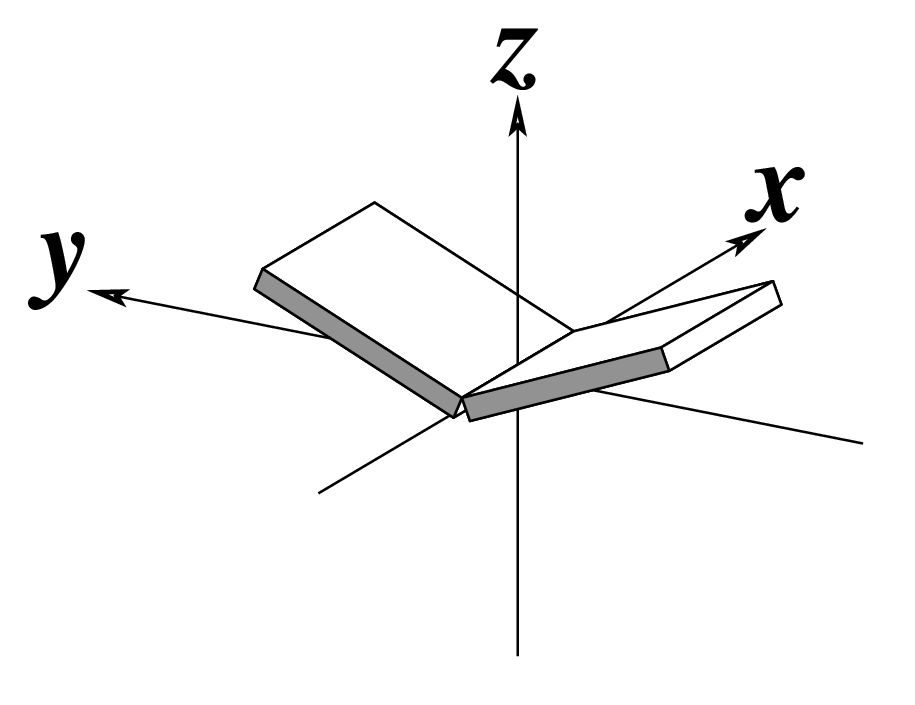 |
--shear | Output shear | Angstroms | 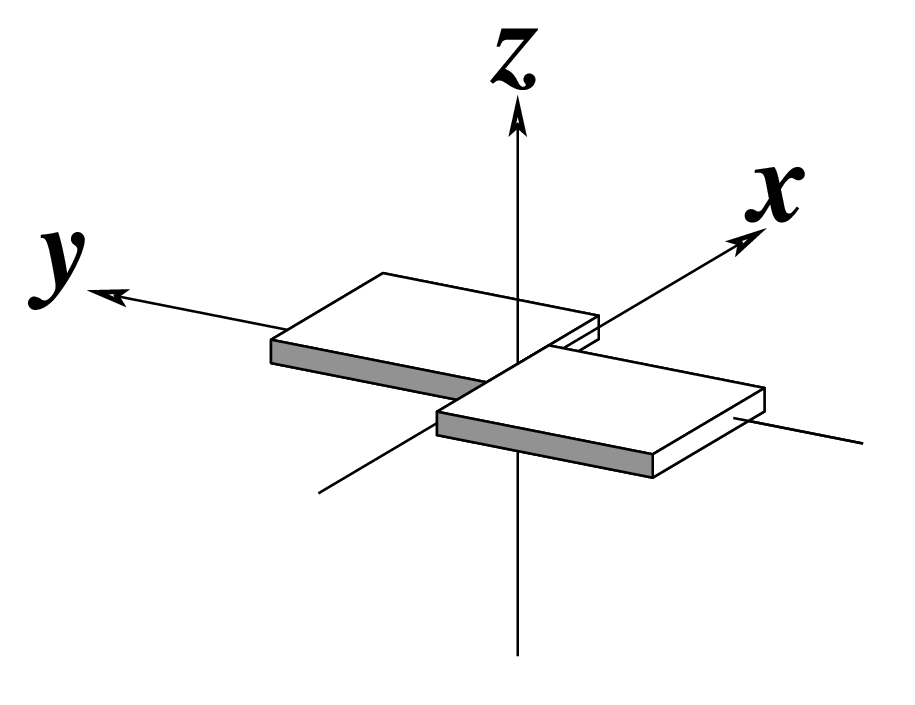 |
--opening | Output opening | Degrees |  |
--stagger | Output stagger | Angstroms | 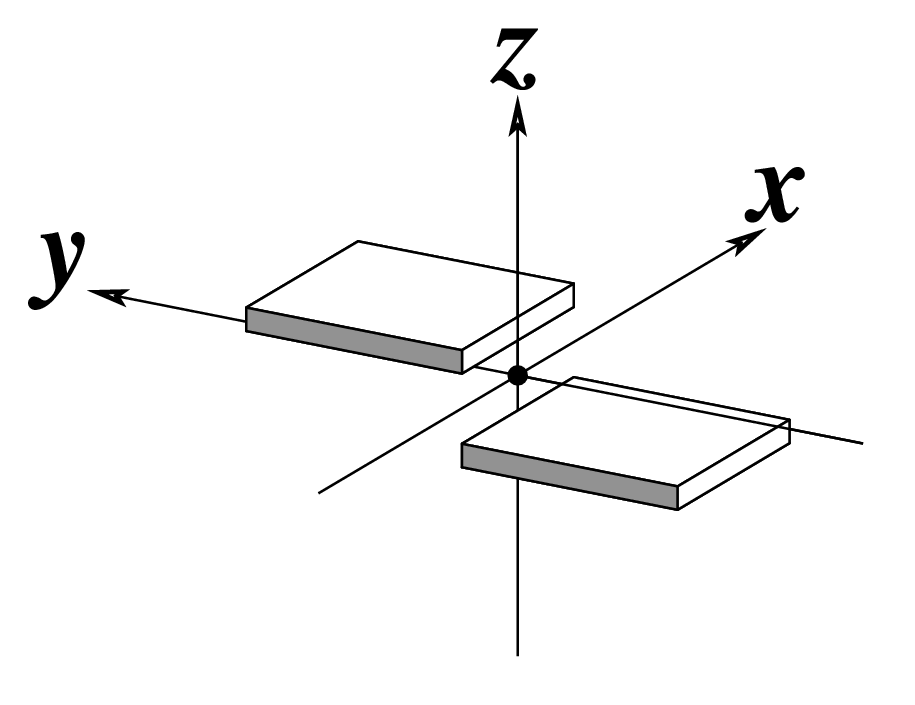 |
--tilt | Output tilt | Degrees | 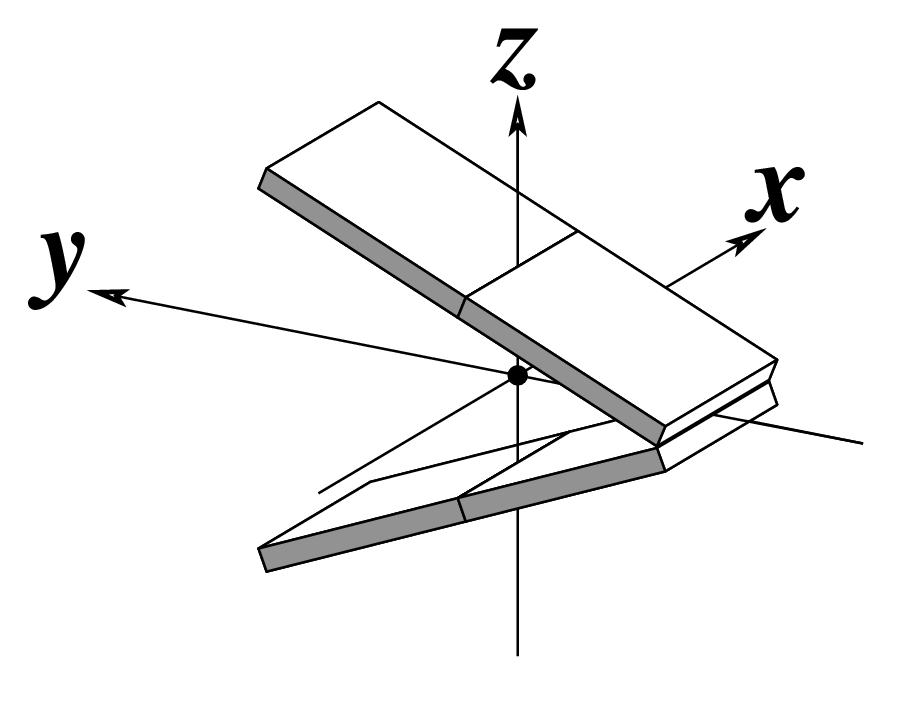 |
--slide | Output slide | Angstroms | 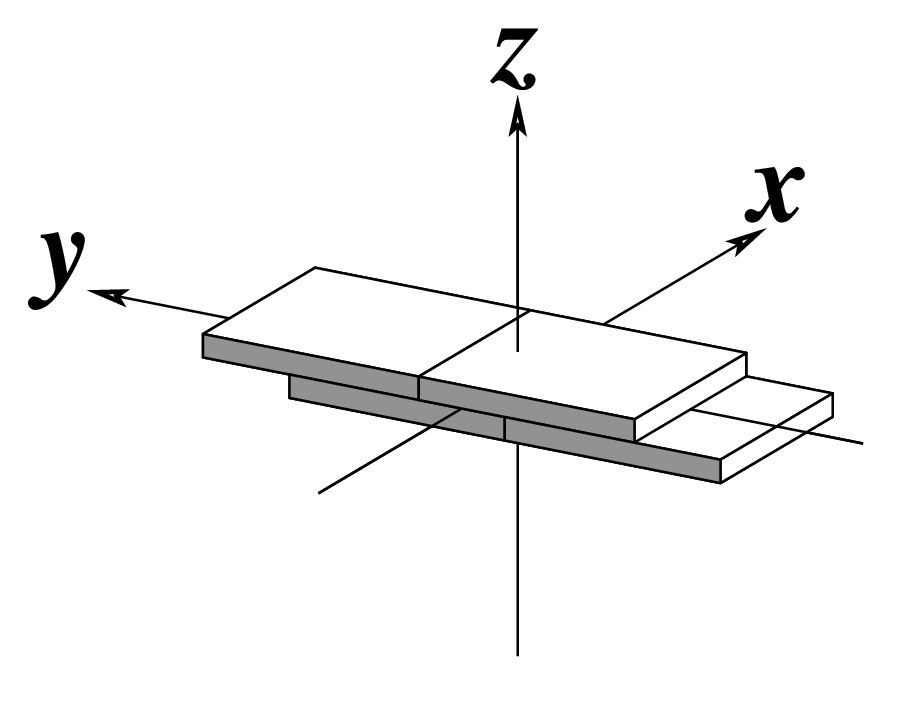 |
--rise | Output rise | Angstroms | 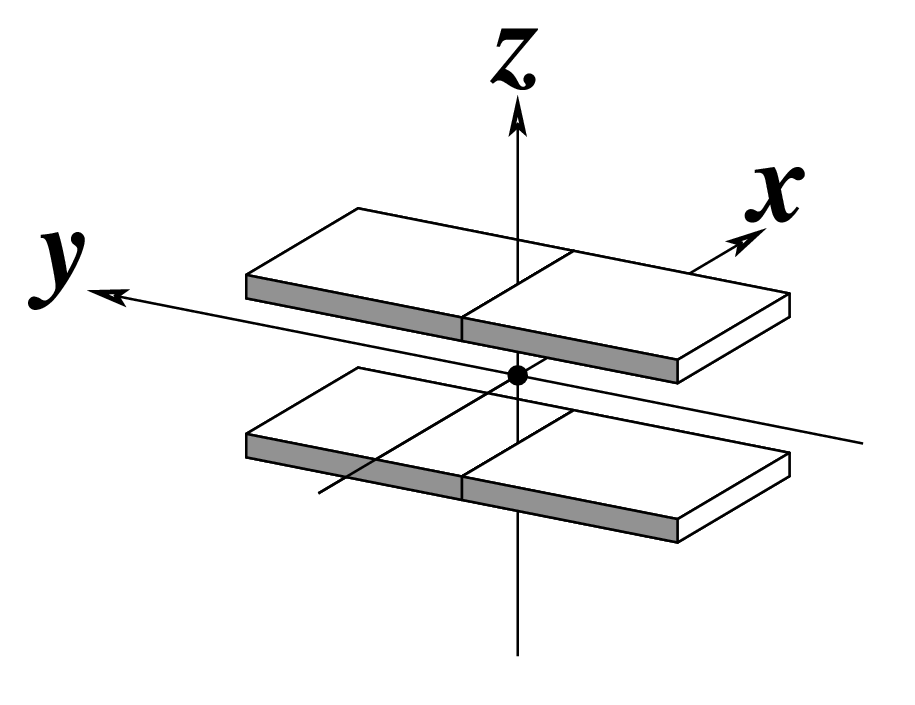 |
--shift | Output shift | Angstroms | 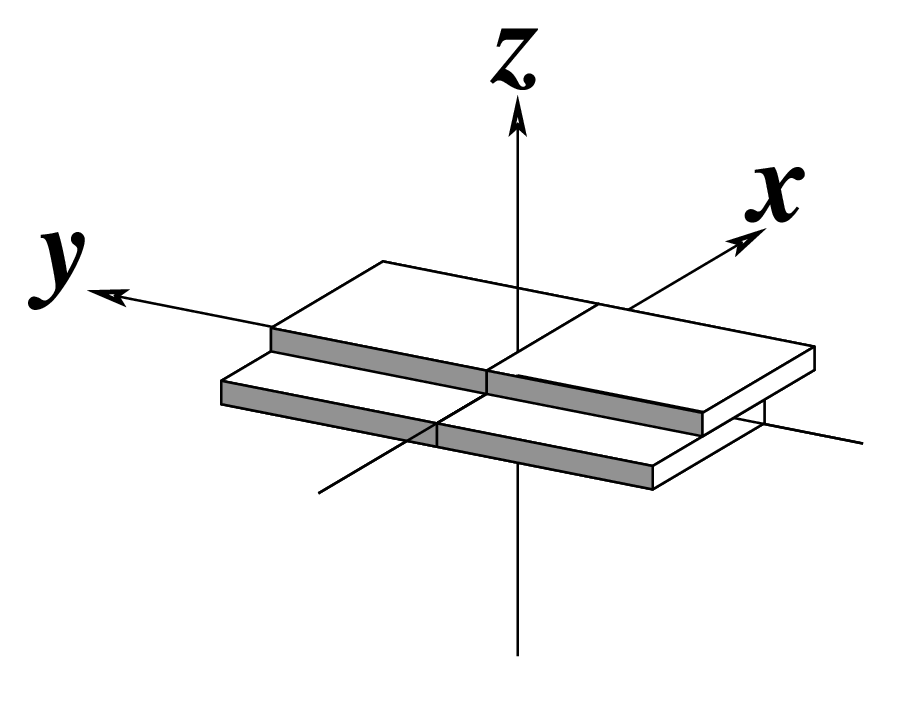 |
--2013 | Output groove, roll, propeller twist, and helical twist (equivalent to -grpl) | ||
-a, --2021 | Output all 14 metrics |
MGW
EP
Base-Pair Schematics