Extract FASTA

Generate FASTA file from indexed Genome FASTA file and BED file. Script will generate FAI index if not present in Genome FASTA folder.
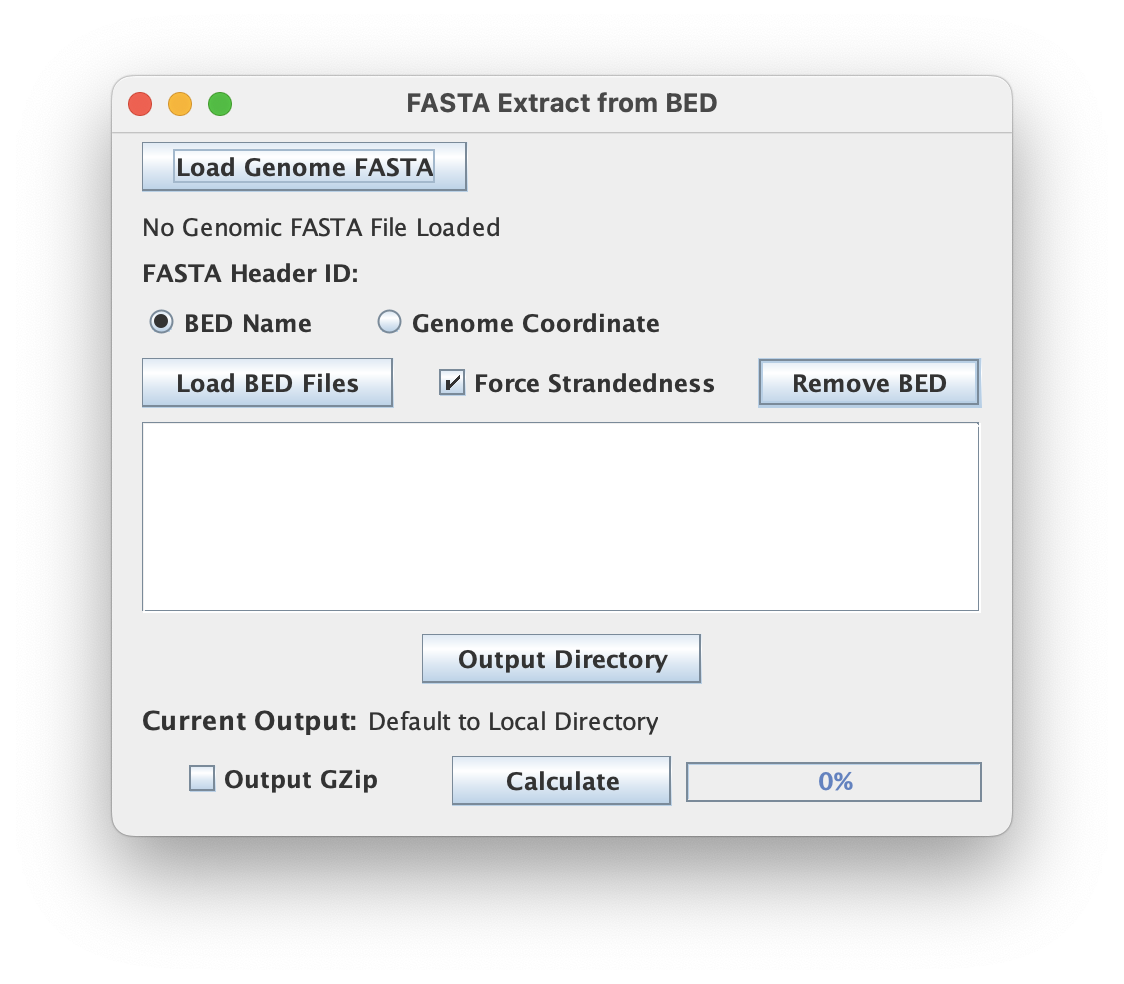
File inputs (Genomic FASTA & BED)
BED files capture coordinate regions without the sequence information. This tool allows the user to search the FASTA file the BED file is based on and extract the sequence within the genomic region to a new FASTA-formatted file. The input FASTA is often a genome FASTA but as long as chrname column matches FASTA identifiers, it could be any FASTA.
When using the GUI, make sure your input is properly formatted and uses the appropriate BED (.bed or .bed.gz) and FASTA (.fa / .fa.gz / .fasta / ...) extensions.
File Options
The 'Force Strandedness' options ensures that the analysis will respect the strand information specified in the BED file when extracting sequences.
Command Line Interface
Usage:
java -jar ScriptManager.jar sequence-analysis fasta-extract [-cfhV] [-o=<output>]
<genomeFile> <bedFile>
Positional Input
The first positional input
| Option | Description |
|---|---|
<fastaFile> | reference genome FASTA file |
<bedFile> | the BED file of sequences to extract |
Output Options
| Option | Description |
|---|---|
-o, --output=<output> | Specify output file |
-z, --gzip | gzip output (default=false) |
Extract Options
| Option | Description |
|---|---|
-c, --coord-header | use genome coordinate for output FASTA header (default is to use bed file headers) |
-f, --force | force-strandedness (default) |