Search Motif

Search for an IUPAC DNA sequence motif in FASTA files with mismatches allowed
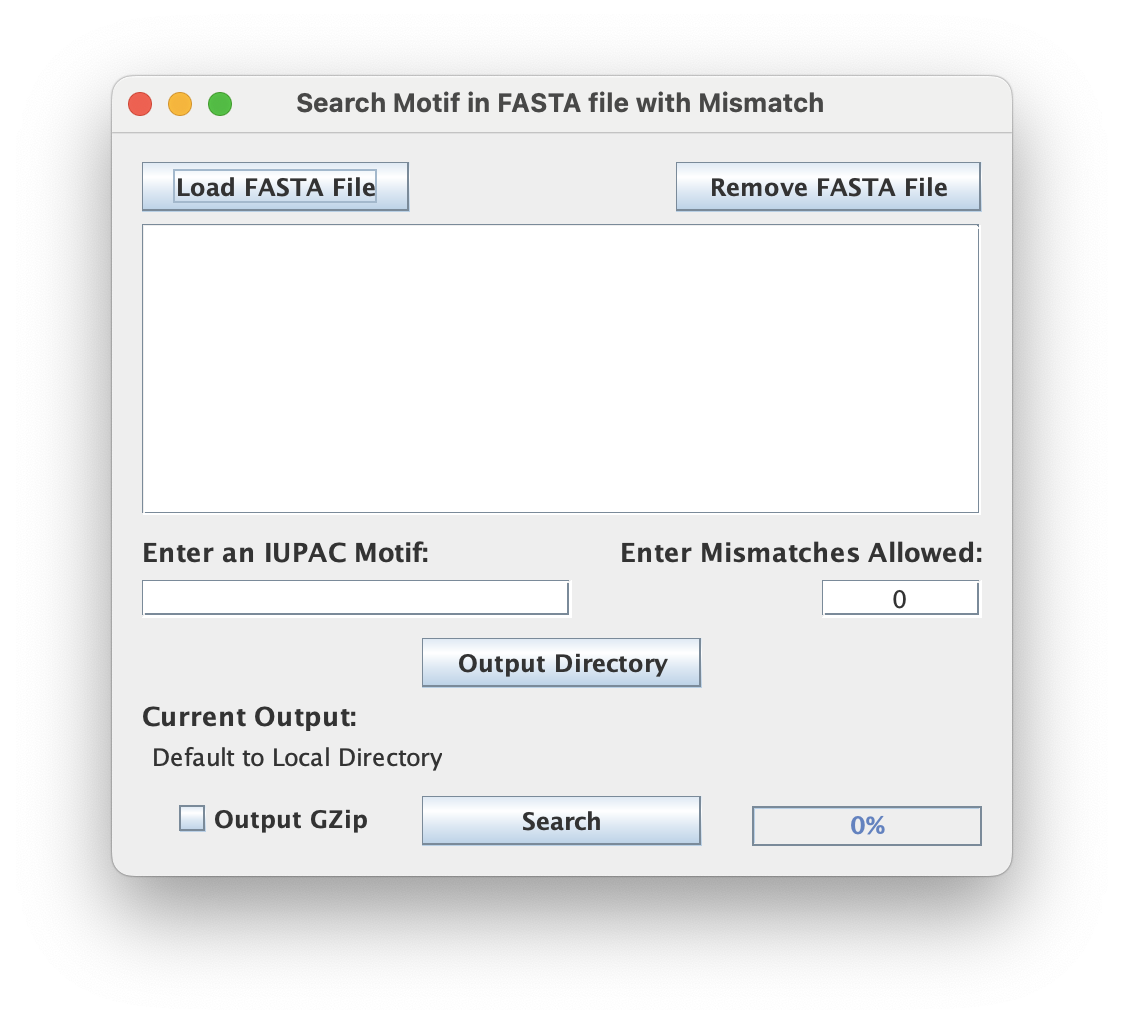
File inputs (FASTA)
Each input FASTA-formatted file will be searched for the user-provided motif. This is typically a genomic FASTA file but can be used with any FASTA formatted file.
When using the GUI, make sure your input is properly formatted and uses the appropriate FASTA (.fa / .fa.gz / .fasta / ...) extensions.
Search Options
Enter an IUPAC Motif
IUPAC (International Union of Pure and Applied Chemistry) has a standard representation for DNA sequences that supports single and sets of bases. Below are some examples but you will need to look up the full IUPAC code for the comprehensive list of options that this tool supports:
- 'A': Adenine
- 'T': Thymine
- 'C': Cytosine
- 'G': Guanine
- 'R': Purine (A or G)
- 'Y': Pyrimidine (C or T)
- 'N': Any Nucleotide (A, T, C, or G)
These are used to define a DNA pattern to search for within the input FASTA sequences.
Enter Mismatches Allowed
The user can toggle the stringency of the motif search by adjusting the number of mismatched nucleotides that can be tolerated when searching for the motif in the FASTA sequences. Mismatches are positions in the sequence where the nucleotide does not match any of the nucleotides represented in the IUPAC motif for that position.
Command Line Interface
Usage:
java -jar ScriptManager.jar sequence-analysis search-motif [-hV] -m=<motif>
[-n=<ALLOWED_MISMATCH>] [-o=<output>] <fastaFile>
Positional Input
| Option | Description |
|---|---|
<fastaFile> | reference genome FASTA file |
Output Options
| Option | Description |
|---|---|
-o, --output=<output> | specify output file |
-z, --gzip | gzip output (default=false) |
Search Options
| Option | Description |
|---|---|
-m, --motif=<motif> | the IUPAC motif to search for |
-n, --mismatches=<ALLOWED_MISMATCH> | the number of mismatches allowed (default=0) |