ChIP-exo Peak Calling Tutorial
Generate peak calling frequency plots from ChIP-exo data
Goal: This tutorial provides a guide to performing a peak calling analysis using the ScriptManager platform and data generated by the Yeast Epigenome project.
Download ScriptManager (v0.14):
The current version of ScriptManager is available for download here. Make sure you have Java installed.
The file ScriptManager-v0.14.jar should be placed someplace locally accessible. For example on Mac OS on the Desktop (Permissions will need to be accepted) or someplace in your home directory (Macintosh_HD:Users/userID/ScriptManager)
Download Python Scripts
This tutorial will use Python scripts GeneTrack and CWPair. Make sure you have Python installed.
(Optional) Download USCS binary bedGraphToBigWig
You will need to download the precompiled binary for converting bedGraphs to BigWig files. Navigate to UCSC executables FTP. Click the link to the OS that matches your machine and scroll down to download the bedGraphToBigWig binary.
- Linux/MacOS
Make sure you modify the permissions so that you can execute the binary. You can do this with chmod
chmod 755 /path/to/bedGraphToBigWig
For example, if your bedGraphToBigWig binary downloaded to your Downloads folder,
chmod 755 ~/Downloads/bedGraphToBigWig
Download Data
You need one set of aligned DNA fragments (BAM) to complete this exercise. Read more about the BED/BAM file formats here.
BAM File
This is the set of Reb1 read alignments from the Yeast Epigenome Project (YEP). See Rossi et al (2021) for more details.
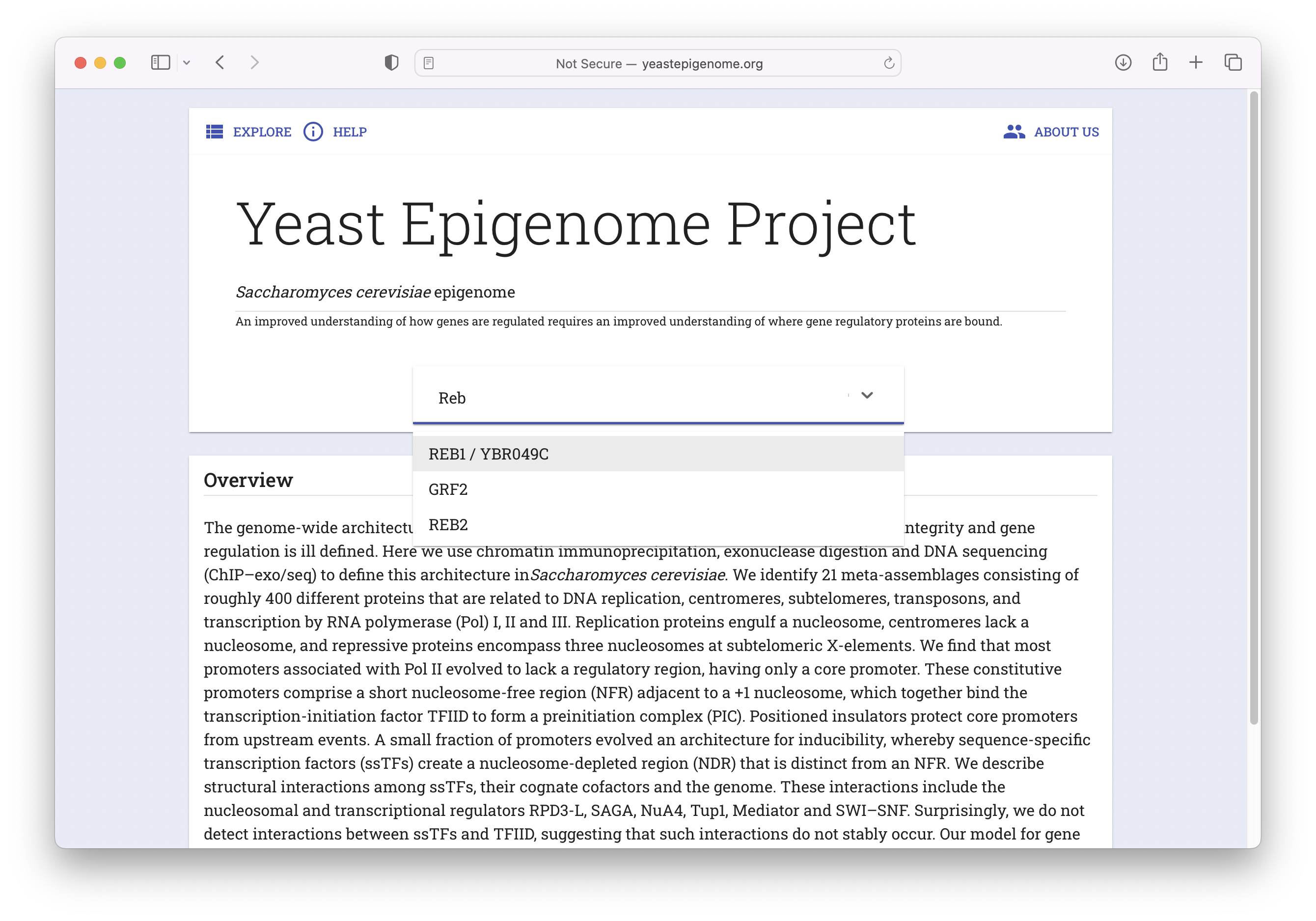
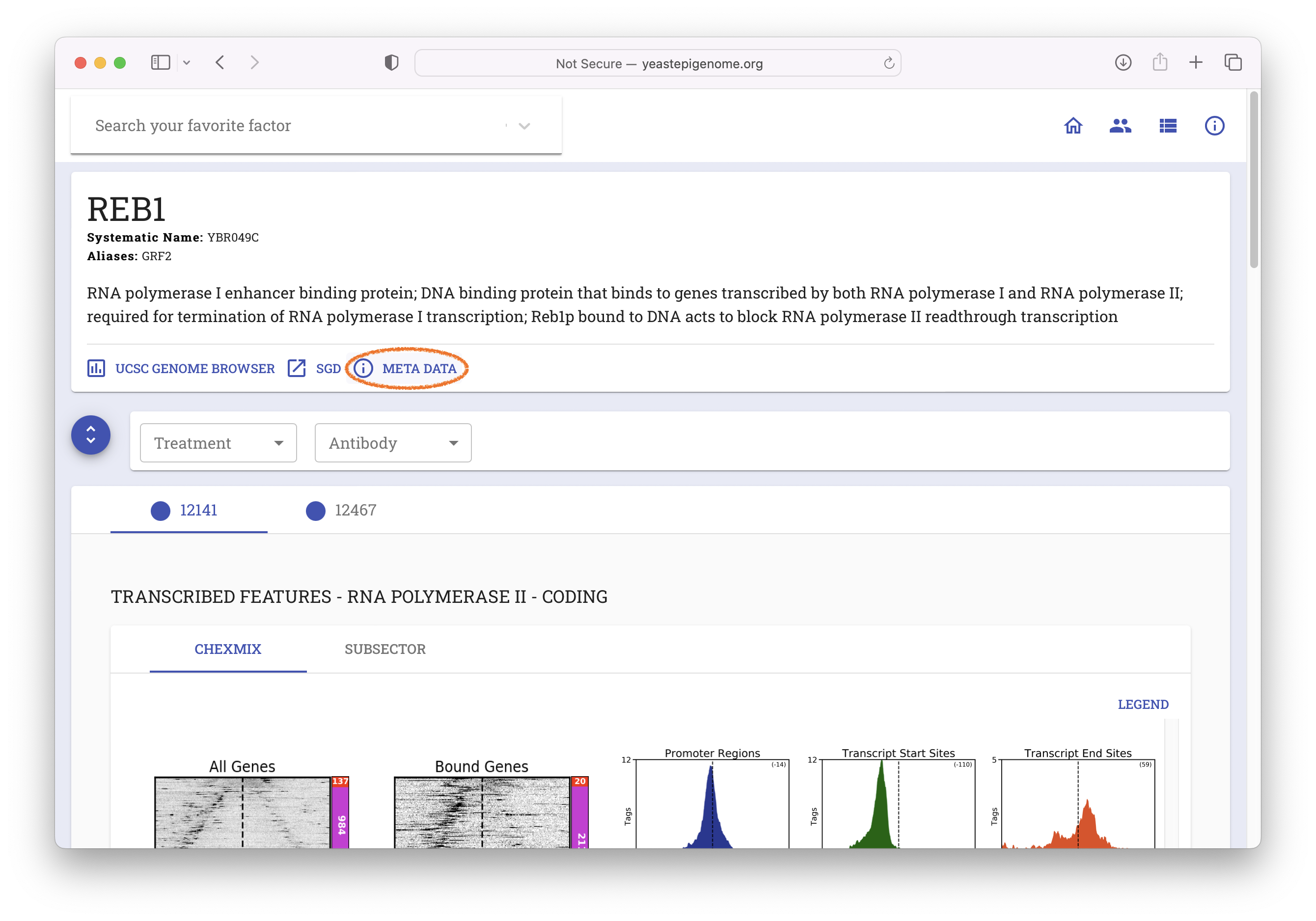
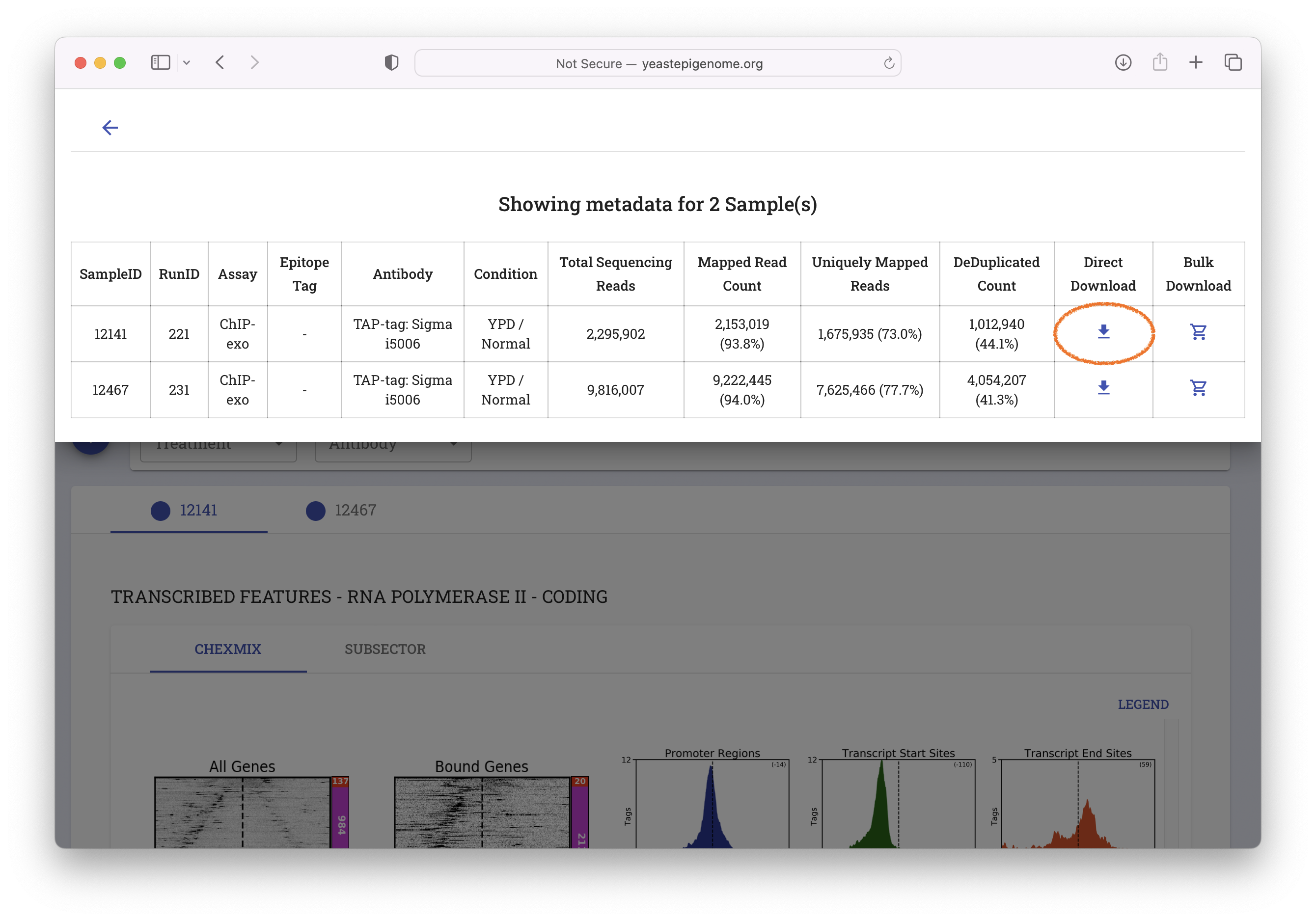
- Navigate to www.yeastepigenome.org and search for Reb1
- Select "META DATA"
- Select "Direct Download"
- Unzip the resulting file ‘12141_YEP.zip’ and inspect the contents of the new
12141_YEPfolder. It should contain a file called12141_filtered.bam.
(Optional) XXXX.chrom.sizes Reference File
You will also need the chromosome sizes file of the yeast reference genome (sacCer3):
Download sacCer3.chrom.sizes (TXT)The downloaded sacCer3.chrom.sizes file may be using the roman numeral naming system (chrI chrII chrIII ...). You will need to modify this to the arabic (chr1 chr2 chr3 ...) numeral naming system if you plan to use the BAM file linked above. You can do this in a simple text editor.
Also, if the file is missing the 2-micron row, you will need to add it manually. Simply append the following tab-delimited row to the bottom of the sacCer3.chrom.sizes file.
2-micron 6318
Convert BAM File to scIDX Format
1. Open ScriptManager
- MacOS
- Linux
- Windows
Depending on your system permissions, you may need to be an administrator to open this for the first time. On Mac systems, this can be done by right-clicking the file and selecting ‘Open’ at the top.
Some MacOS systems may not properly open the JAR by simply double-clicking on the JAR file so you may need to open your Terminal window and execute it from the command line by executing the jar file without any arguments or flags:
java -jar /path/to/ScriptManager.jar
If you're not sure how to type the path to ScriptManager, you can type java -jar (end with space) and then drag ScriptManager from Finder into your Terminal window and then press enter.
Double-click or right-click the ScriptManager JAR file to start the program.
Double-click or right-click the ScriptManager JAR file to start the program.
Once you see the main tool selection window, you're off to the races!
2. Generate BAI index file
A BAI index file is required for each BAM file of interest (i.e., the tag occupancy data you want to plot). This file allows for rapid access of the sorted and aligned sequence reads (BAM file).
SAM/BAM standard is to keep BAI file in same directory as BAM file with the ScriptManager-generated filename.
Reb1_YEP_12141.bam
Reb1_YEP_12141.bam.bai # Need to generate this file to proceed.
2.1. Navigate to BAM Manipulation ➡️ BAM-BAI Indexer
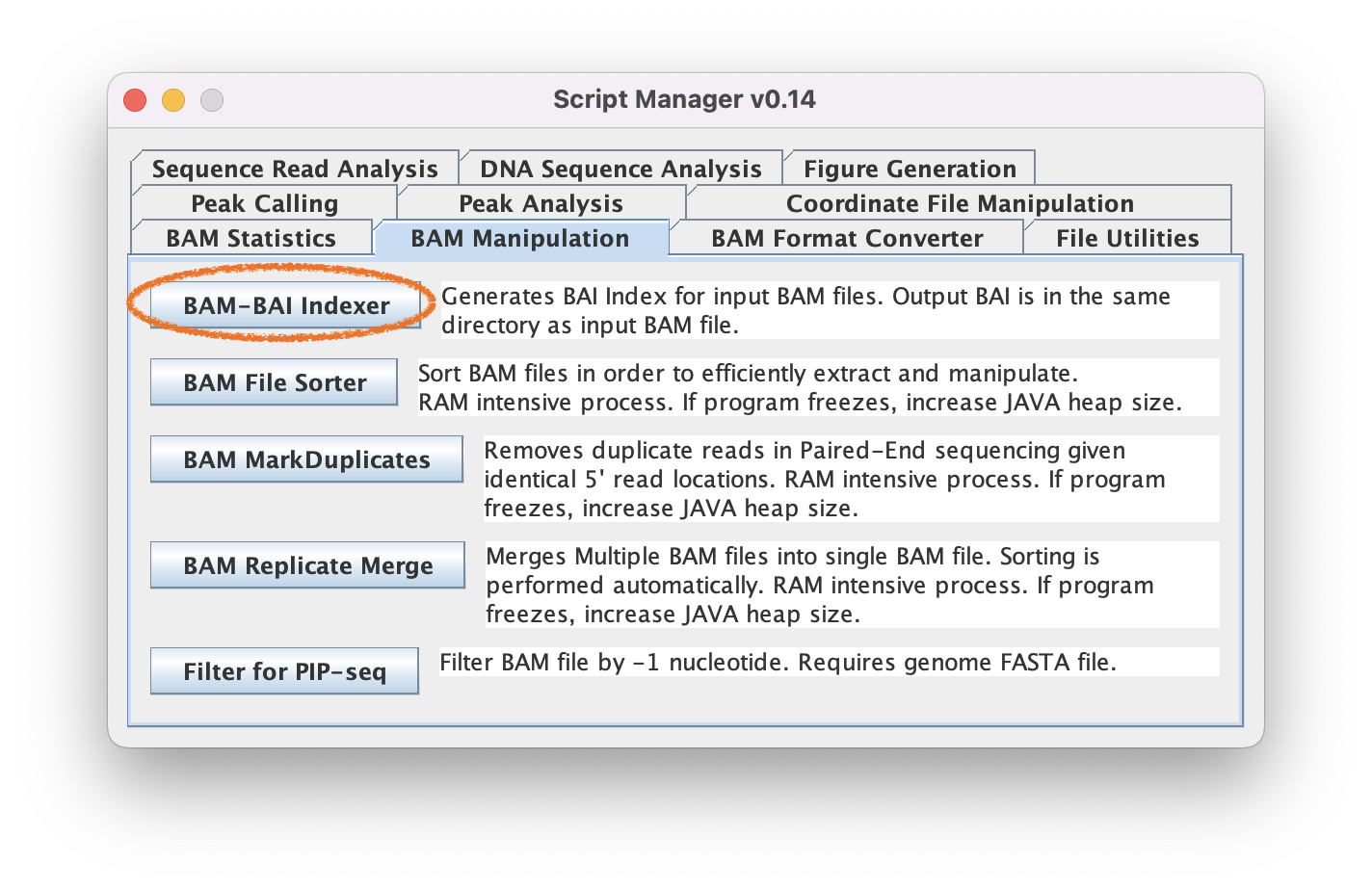
2.2. Generate BAI index files for each BAM file of interest by loading your BAM file and clicking "Index."
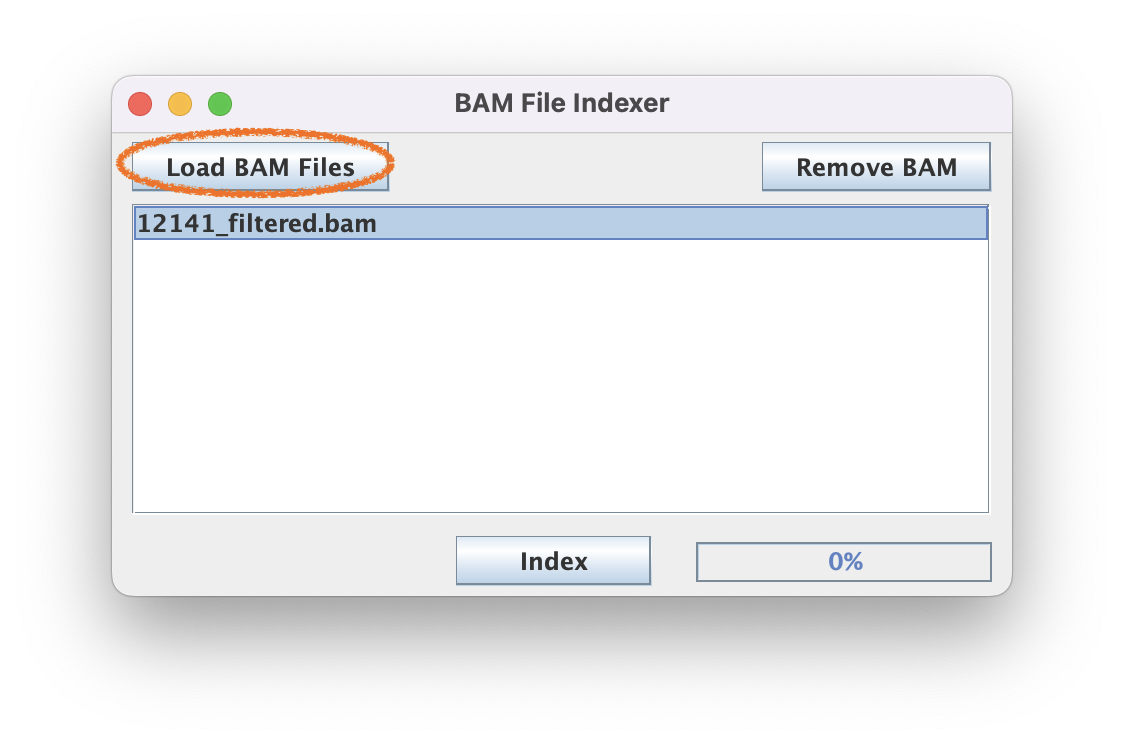
The speed of this step scales with the size of the BAM file. Generally this step 30 sec for a 100 MB BAM file but may take 1-2 min for a multi-GB BAM file.
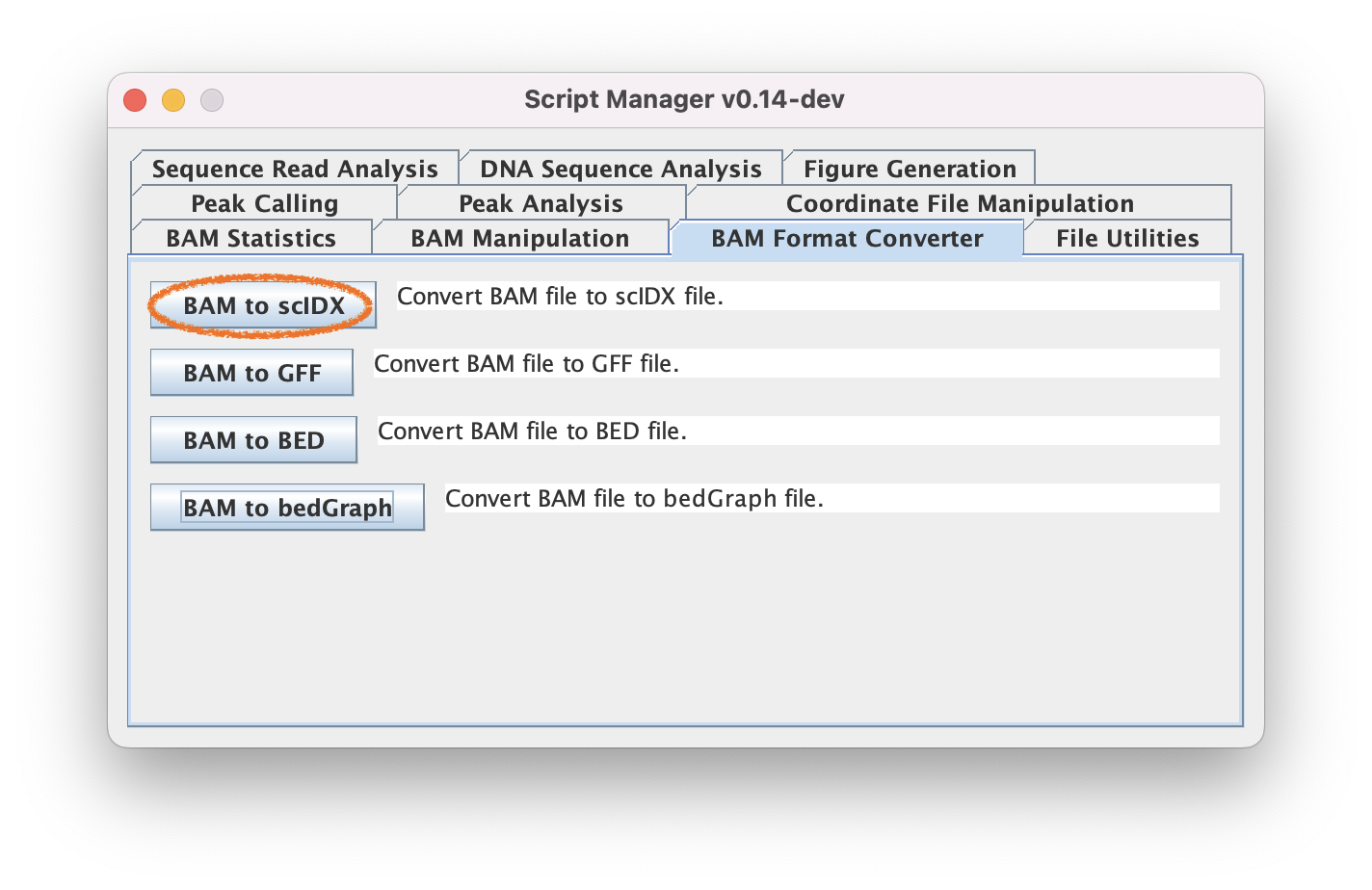
3. Create a whole-genome pileup using BAM Format Converter Tools
2.1. Navigate to BAM Format Converter ➡️ BAM to scIDX to do a genome-wide pileup of your BAM
2.2. Select Encoding information
Since this is a ChIP-exo dataset, we will select Read 1 (5' end) for the encoding. You may select insert size filters if you wish.
2.3. Execute pileup
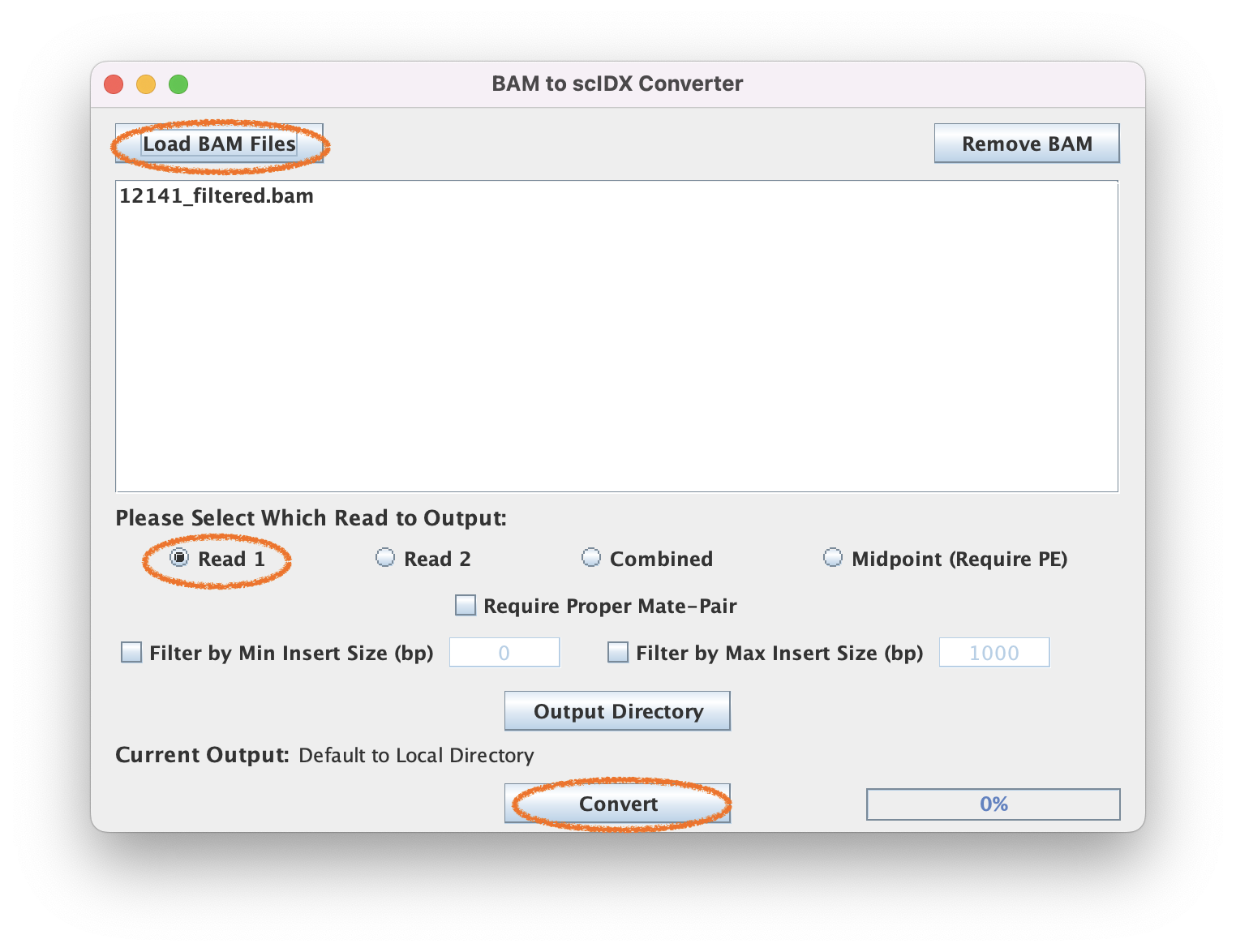

This should output a 12141_filtered_READ1.tab file.
GeneTrack Script
Open the terminal on your computer. Navigate to the directory with the genetrack.py file and run the following command:
python genetrack.py /path/to/12141_filtered_READ1.tab
Output called peaks in GFF format
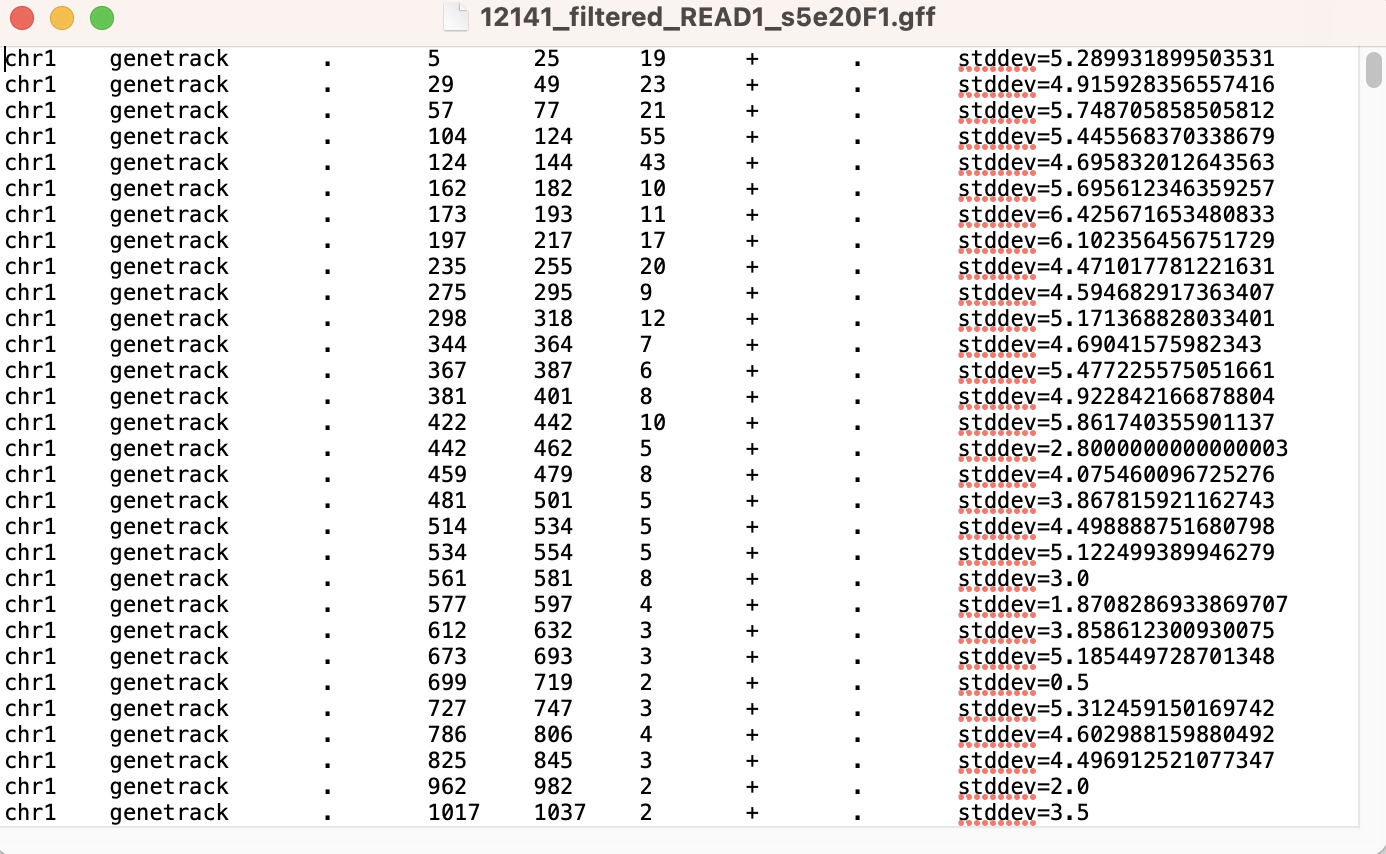
This should output a folder labeled genetrack_s5e20F1 in the directory of your scIDX file. Inside of the folder, the 12141_filtered_READ1_s5e20F1.gff file will be used for the CWPair script.
The GeneTrack script supports multiple input files. You can run the script using a file or list of files, a directory or list of directories, or use "." to run the script in the current directory.
Output Options
| Option | Description |
|---|---|
-s=<SIGMA> | Sigma to use when smoothing reads to call peaks (default=5) |
-e=<EXCLUSION> | Exclusion zone around each peak that prevents others from being called (default=20) |
-u=<UP_WIDTH> | Upstream width of called peaks (default=half exclusion zone) |
-d=<DOWN_WIDTH> | Downstream width of called peaks (default=half exclusion zone) |
-F=<FILTER> | Absolute read filter; outputs only peaks with larger read count (default=1) |
CWPair Script
Now that the GeneTrack script output has been generated, we can run the CWPair script. Navigate to the directory with the cwpair.py file and run the following command:
python cwpair.py /path/to/12141_filtered_READ1_s5e20F1.gff
This should output a folder labeled cwpair_output_mode_f0u50d100b1 inside of the previous genetrack_s5e20F1 folder. This folder contains the following files:
S_12141_filtered_READ1_s5e20F1.gff (simple),
D_12141_filtered_READ1_s5e20F1.txt (detailed),
O_12141_filtered_READ1_s5e20F1.txt (orphans),
P_12141_filtered_READ1_s5e20F1.pdf (frequency preview plot),
F_12141_filtered_READ1_s5e20F1.pdf (final frequency plot),
statistics.txt (summarizes each input file)
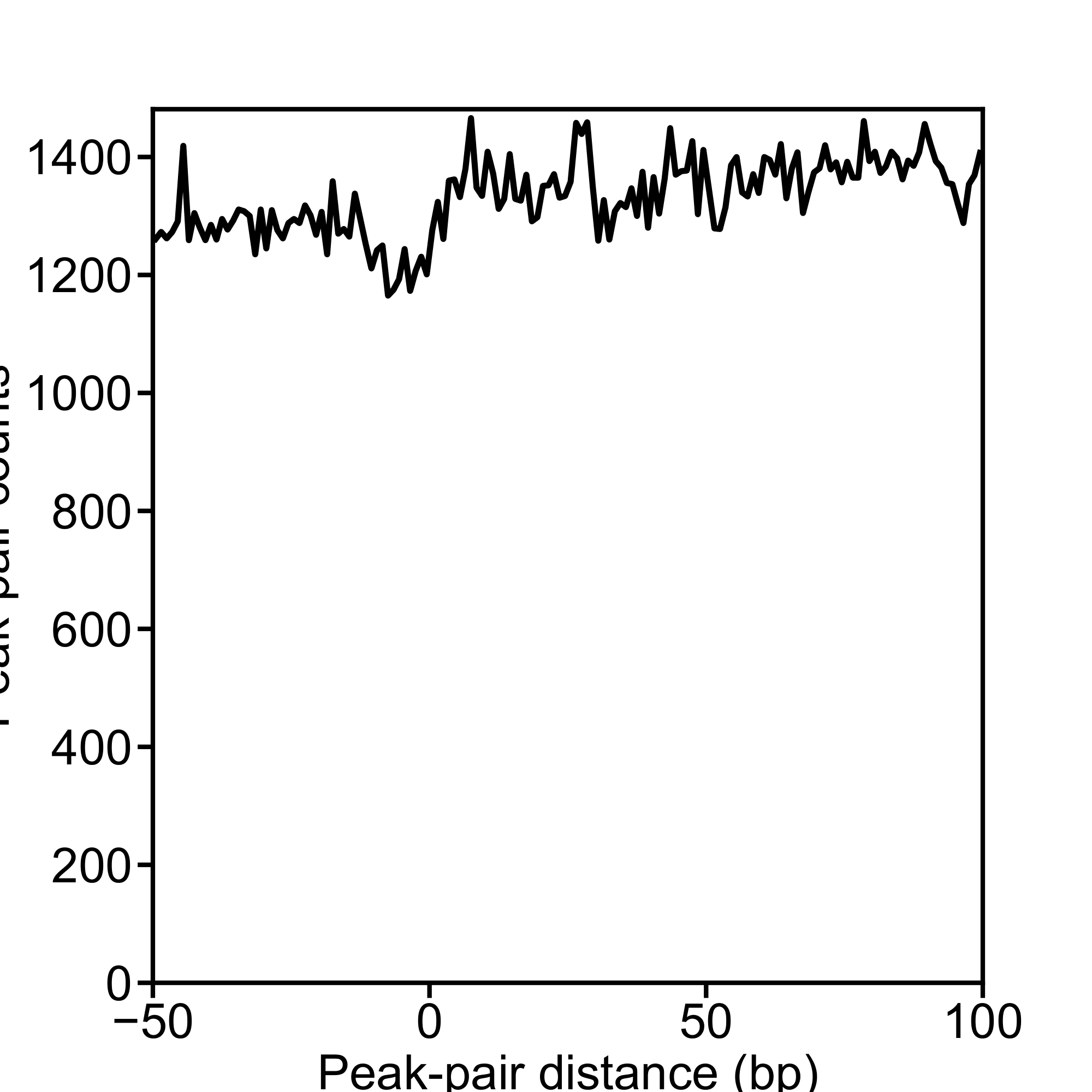
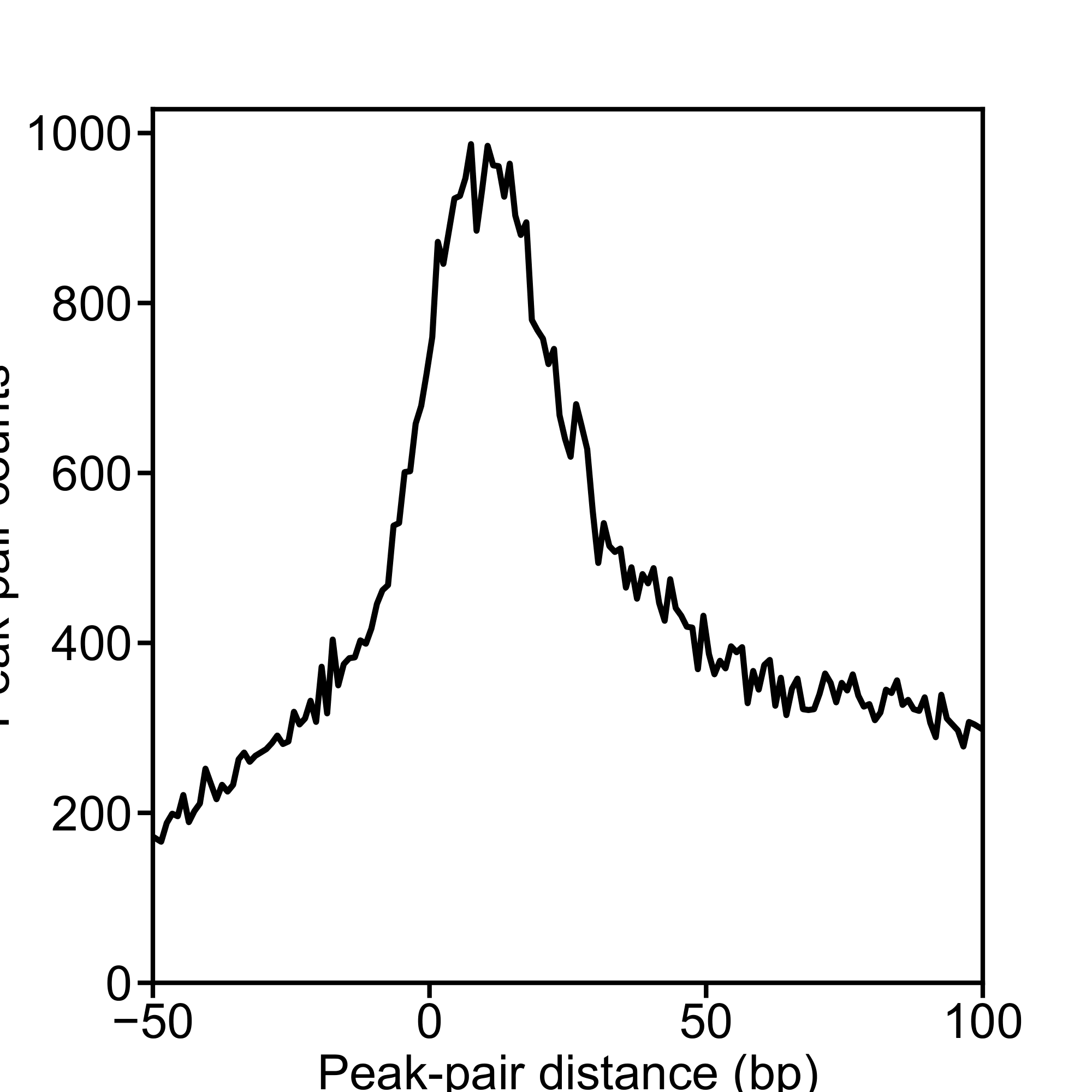
.
The CWPair script supports multiple input files. You can run the script using a file or list of file, a directory or list of directories, or use "." to run the script in the current directory.
Output Options
| Option | Description |
|---|---|
-m=<METHOD> | Method of finding match. Valid options are mode (default), closest, largest, and all (run with each method) |
-u=<UP_DISTANCE> | Distance upstream of a Watson peak to allow a Crick pair (default=50) |
-d=<DOWN_DISTANCE> | Distance downstream of a Watson peak to allow a Crick pair (default=100) |
-d=<BINSIZE> | Width of bins for frequency plots and mode calculation (default=1) |
-F=<ABSOLUTE_THRESHOLD> | Absolute read filter; outputs only peaks with larger read count (default=0) |
-p=<PLOT_FORMAT> | Format to output graph in. Valid options are png, svg, and pdf (default) |
-o=<FORMAT> | Format to output simple output file in. Valid options are gff (default) and txt |
-c=<SORT_CHROMOSOME> | Output files will be sorted by chromosome. Valid options are asc (default) and desc |
-s=<SORT_SCORE> | Output files will be sorted by score. Valid option are asc, desc, and none (default) |
(Optional) View Pileup in Genome Browser
Follow these steps to turn your BAM files into genome track files that can be viewed in a genome browser!
Convert scIDX to BigWig
Since ScriptManager currently does not support converting BedGraph files to BigWigs, this step must be done in your terminal/on the command-prompt.
1. Turn your scIDX file into a bedGraph file
- Shell
- Excel
grep -v -f sacCer3.chrom.sizes 12141_filtered_READ1_SCALE.tab > 12141_filtered_READ1_SCALE.filtered.tab
sed '1d;2d' 12141_filtered_READ1_SCALE.filtered.tab | awk '{OFS="\t"}{FS="\t"}{print $1, $2, $2+1, $3}' |sort -k1,1 -k2,2n > 12141_filtered_READ1_SCALE_forward.bedgraph
sed '1d;2d' 12141_filtered_READ1_SCALE.filtered.tab | awk '{OFS="\t"}{FS="\t"}{print $1, $2, $2+1, $4}' |sort -k1,1 -k2,2n > 12141_filtered_READ1_SCALE_reverse.bedgraph
Open your file in excel and strip the file of the first two rows:
#2023-03-10 12:52:40.439;12141_filtered.bam;READ1
chrom index forward reverse value
You may also have read mapping to the very edges of the chromosome. These entries will also have to be removed.
Then rearrange the columns into BedGraph format by copying them in the following order (3rd column is the value of col2 plus one):
12141_filtered_READ1_SCALE_forward.bedGraph: col1, col2, (col2 +1), col312141_filtered_READ1_SCALE_reverse.bedGraph: col1, col2, (col2 +1), col4
...and then sort each of them by the first and then the second columns. This format is tab-delimited (not comma-delimited!) so save as a tab-delimited text file. Do not save this as an Excel file format (.xlsx, .xls, etc). This should be a .tsv or .tab format.
2. Turn your bedGraph file into a BigWig file
- Shell
./bedGraphToBigWig 12141_filtered_READ1_SCALE_forward.bedGraph sacCer3.chrom.sizes my-track-basename_forward.bw
./bedGraphToBigWig 12141_filtered_READ1_SCALE_reverse.bedGraph sacCer3.chrom.sizes my-track-basename_reverse.bw
3. Load the BigWig into your favorite genome browser
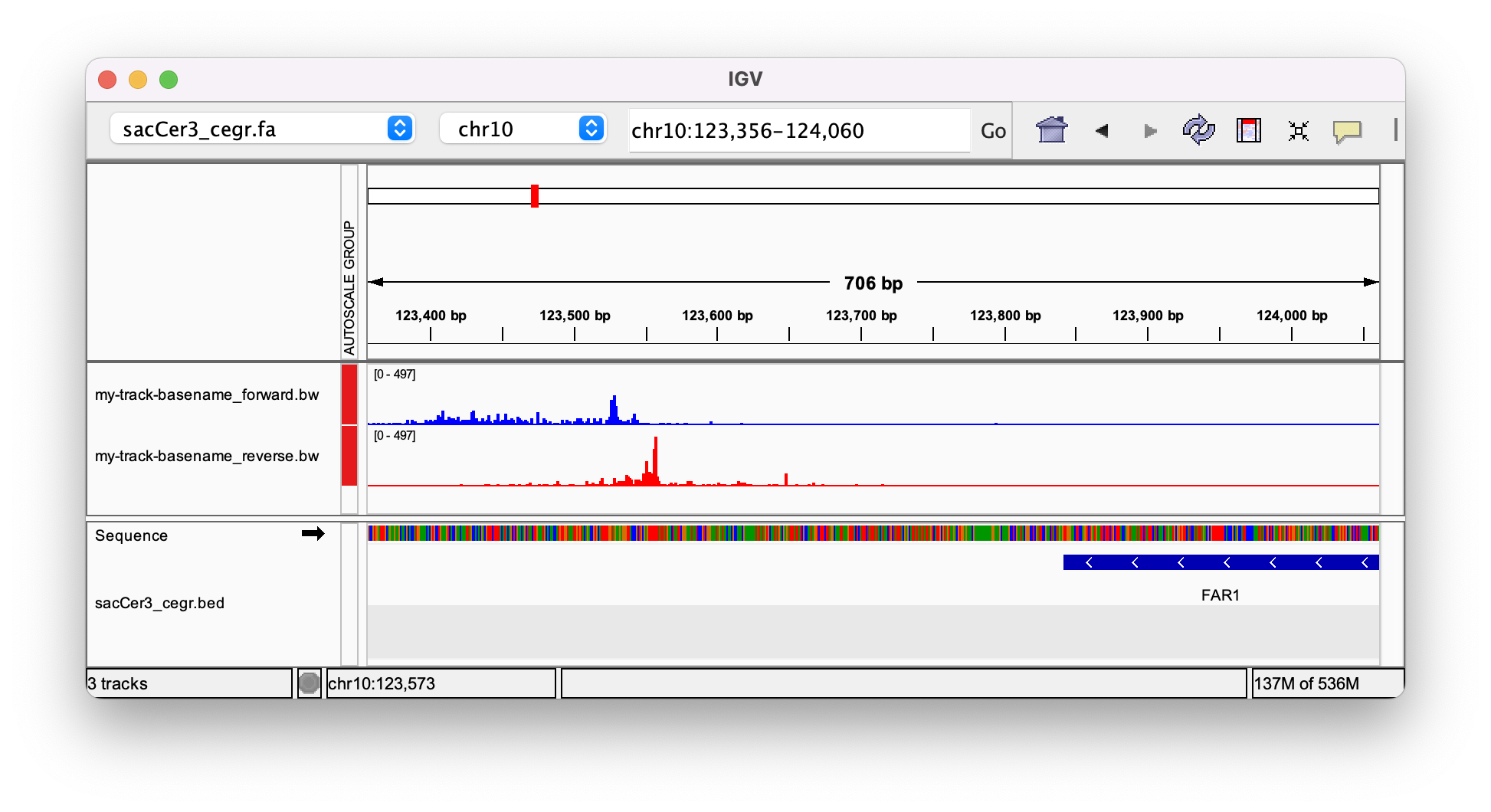
Tah dah! You've made the genome tracks for a ChIP-exo dataset!
Command-Line shell script
SCRIPTMANAGER=/path/to/ScriptManager.jar
BGTOBW=/path/to/bedGraphtoBigWig
BAMFILE=/path/to/12141_filtered.bam
OUTPUT=/path/to/output-basename
CHRMSZ=/path/to/sacCer3.chrom.sizes
GENETRACK=/path/to/genetrack.py
CWPAIR=/path/to/cwpair.py
# Index BAM if not already indexed
[ -f $BAMFILE.bai ] || samtools index $BAMFILE
# Pileup BAM along whole genome
java -jar $SCRIPTMANAGER bam-format-converter bam-to-scidx $BAMFILE -o $OUTPUT.tab
# Run GeneTrack script
python $GENETRACK $OUTPUT.tab
# Run CWPair script
python $CWPAIR $OUTPUT_s5e20F1.gff
# (Optional) View pileup in genome browser
# Only include pileups from chromosomes in the sacCer3.chrom.sizes reference
grep -v -f $CHRMSZ $OUTPUT.tab > $OUTPUT.filtered.tab
# Convert to forward/reverse Bedgraph tracks
sed '1d;2d' $OUTPUT.scaled.tab | awk '{OFS="\t"}{FS="\t"}{print $1, $2, $2+1, $3}' |sort -k1,1 -k2,2n > $OUTPUT.forward.bedgraph
sed '1d;2d' $OUTPUT.scaled.tab | awk '{OFS="\t"}{FS="\t"}{print $1, $2, $2+1, $4}' |sort -k1,1 -k2,2n > $OUTPUT.reverse.bedgraph
# Compress to BigWig format
$BGTOBW $OUTPUT.forward.bedgraph $CHRMSZ $OUTPUT\_forward.bw
$BGTOBW $OUTPUT.reverse.bedgraph $CHRMSZ $OUTPUT\_reverse.bw
# Clean-up
rm $OUTPUT.tab $OUTPUT.filtered.tab $OUTPUT.forward.bedgraph $OUTPUT.reverse.bedgraph
# Output files:
# - /path/to/S_output-basename_s5e20F1.gff
# - /path/to/D_output-basename_s5e20F1.txt
# - /path/to/O_output-basename_s5e20F1.txt
# - /path/to/P_output-basename_s5e20F1.pdf
# - /path/to/F_output-basename_s5e20F1.pdf
# - /path/to/my-track-basename_forward.bw (optional)
# - /path/to/my-track-basename_reverse.bw (optional)